










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Reumatología
versión On-line ISSN 1817-5996
Rev Cuba Reumatol vol.16 no.3 La Habana sep.-dic. 2014
ARTÍCULO DE REVISIÓN
Enfermedad de Behçet
Behcet's disease
William Castillo González I, Javier González-Argote I, Jorge Hernández Estévez I
I Hospital Dr. Miguel Enríquez. Laboratorio Central de Líquido Cefalorraquídeo (LABCEL). Facultad de medicina Dr. Miguel Enríquez. Universidad de Ciencias Médicas de La Habana. La Habana, Cuba.
RESUMEN
La Enfermedad de Behçet es una enfermedad inflamatoria crónica, recurrente, multisistémica, de etiología desconocida. Todas las manifestaciones son autolimitadas excepto la afección ocular. Se caracteriza por períodos de remisión y de exacerbación de frecuencia y duración impredecibles. El diagnóstico se basa en la asociación de aftosis bipolar, manifestaciones cutáneas, uveítis, sobre todo posterior y afectación de grandes vasos. En ausencia de estos signos es más difícil confirmar el diagnóstico. La afección del tracto gastrointestinal y sistema nervioso central es menos frecuente, aun así pueden amenazar la vida del paciente. La susceptibilidad de la enfermedad de Behçet está fuertemente asociada con la presencia del alelo HILA-B51. Las manifestaciones neurológicas suelen ser signos de malignidad.
Palabras clave: enfermedad de Behçet, síndrome de Behçet, úlceras orales, úlceras genitales, uveítis.
ABSTRACT
The Behçet's disease is a chronic inflammatory disease, recurrent, multisystem of unknown etiology. All events are self-limited except the ocular condition. It characterized by periods of exacerbation remission and unpredictable frequency and duration. Is based the diagnosis is based on the association of especially bipolar aphthosis, skin manifestations, uveitis and affectation of great vessels. In the absence of these signs is more difficult to confirm the diagnosis. The lesions of the gastrointestinal tract and central nervous system is less frequent, even so may threaten the patient's life. The susceptibility of Behçet´s disease is strongly associated with the presence of HILA -B51 allele. The neurological manifestations are signs of malignancy.
Keywords: disease Behçet, Behçet syndrome, oral ulcers, genital ulcers, uveitis.
INTRODUCCIÓN
A casi un cuarto de siglo de la primera vez que en las revistas científicas cubanas apareciera descrito un caso de Enfermedad de Behçet (EB), mucho se ha investigado en referente a esta enfermedad.1
Al traste con lo anteriormente expuesto poco se ha profundizado en las manifestaciones neurológicas. La presente revisión pretende recoger de manera clara y precisa cuanto se ha avanzado en el estudio de esta importante correlación.
La EB fue descrita por el dermatólogo turco Hulusi Behçet en 1937 estableciendo la triada característica de perturbaciones del ojo, úlceras genitales y orales recurrentes.2
En 1990 se establecen por primera vez los criterios para esta entidad por el International Study Group for Behçet's Disease definiendo la existencia de ulceración oral recurrente con al menos dos de las siguientes manifestaciones clínicas: ulceración genital recurrente, afectación ocular, afectación cutánea o test de patergia positivo para hacer el diagnóstico.3-5

Posteriormente fueron revisados dando lugar a los criterios del 2006,[Tabla 1] que incluye las trombosis arteriales, venosas, y aneurismas de grandes vasos, teniendo mayor especificidad con esta propuesta.6
Afecta principalmente a los adultos jóvenes, y es considerad una enfermedad autoinmune pues su principal lesión antinómico-patológica es una vasculitis.5,6
El tratamiento, con frecuencia poco satisfactorio, es esencialmente sintomático relacionado con el predominio de las manifestaciones clínicas. Al planificarlo hay que tener en cuenta que la enfermedad cursa con remisiones y exacerbaciones impredecibles, el mal pronóstico cuando comienza a edad temprana y en el sexo masculino, así como la afectación de órganos vitales.6
Las causas de muerte siguen siendo la ruptura de aneurismas, la enfermedad severa del sistema nervioso central (SNC) y la perforación intestinal, resultando imprescindible identificar de manera precoz estas complicaciones para aplicar las medidas terapéuticas adecuadas.6
OBJETIVOS
Describir los aspectos esenciales y actuales de antecedentes, definición, clasificación, rasgos clínicos, patogénesis, etiología, tratamiento, evolución y pronóstico de la EB.
Describir las manifestaciones neurológicas de la EB.
MÉTODOS
Estrategia de búsqueda de información
Se realizó una revisión bibliográfica en PubMed y SciELO regional. Se encontraron, con el descriptor Behçet´s disease, en PubMed 2284 artículos y con la enfermedad de Behçet en SciELO 1007 estudios, todos publicados en el periodo comprendido entre el 1974 y el 2013. En una segunda etapa se seleccionaron contribuciones que trataran aspectos como antecedentes, definición, clasificación, rasgos clínicos, patogénesis, etiología, tratamiento, evolución y pronóstico. Se eliminaron los artículos duplicados en ambas búsquedas. Finalmente se incluyeron 84 publicaciones útiles para nuestros objetivos.
DESARROLLO
Hulusi Behçet fue un famoso dermatólogo turco, quien ganó una posición importante en la historia médica tras publicar tres casos de la EB en 1937.1,2
Nació en Estambul el 20 de febrero de 1889. Comenzó los estudios de medicina en la Escuela Médica Militar de Kuleli, en 1901, nueva años después, en 1910, se gradúa de médico y comienza a trabajar de ayudante en el departamento de la dermatología de Gülhane del Hospital Militar, con los eminentes profesores Eqsref Ruqsen, Talat Çamlž, y Resat Ržza.1,2
En agosto de 1918 se vinculó como ayudante voluntario de Blumenthal y Cherevsesky en el Hospital de Charité, primero en Budapest y luego en Berlín.1,2
En octubre de 1919, regresó a Turquía como dermatólogo, ocupando el puesto de jefe de personal del hospital Hasköy dedicado al estudio de las enfermedades venéreas. Posteriormente se traslada al Hospital Vakžf Guraba en Estambul, como director del departamento universitario de dermatología y enfermedades venéreas de Estambul, donde adquiere la categoría profesor donde recibe el título de Profesor distinguido en 1939. Fallece el 8 de marzo de 1948 a la edad de 59 años de una afección cardiaca.1,2
Los estudios médicos de Behçet
La entidad nosológica conocida como "enfermedad de Behçet", ha recibido diferentes sinonimias a través de tiempo estas son: síndrome de Behçet, tríada de Behçet, Tri síntoma que Behçet, La maladie de Behçet o Morbus Behçet, descrita por primera vez en 1924, por el dermatólogo turco Hulusi Behçet.
Después de sus cuidadosas observaciones, decidió que los síntomas que él había reconocido eran los síntomas de una sola enfermedad. Realmente, Hulusi Behçet y muchos otros turcos y doctores de Alemania que trabajan en Turquía estaban haciendo las investigaciones en esta enfermedad.1,2
Sus primeras observaciones clínicas en esta enfermedad las realizó en una paciente que presentaba perturbaciones oculares acompañadas de ulceras orales y genitales, y había sido evaluada en Estambul y Viena durante varios años, con los diagnósticos presuntivos de tuberculosis, sífilis, o un microorganismo que no estaba presente en Europa. Hulusi Behçet continuó examinando pacientes que le fueron remitidos por signos y síntomas similares, pensando que el agente causal era un virus.
Continúo sus estudios sobre virología con el apoyo de Hugo Braun, importante investigador en el campo de la microbiología. Cuando los enfermos con signos similares en Turquía alcanzo el número de cinco en el año 1936, Hulusi Behçet hizo su primer reporte con los datos acumulado en el "Journal of Skin and Veneral Diseases", más tarde en la Sociedad Dermatológica de París, y en otros centros investigativos de Europa.3
El nombre de la enfermedad se sugirió en septiembre de 1947, en el transcurso del Congreso Internacional de Dermatología de Ginebra, donde a pedido del Profesor Miescher y la aprobación de los participantes del congreso, se denominó a la enfermedad caracterizada por úlceras genitales y orales recurrentes asociada a manifestaciones oculares "morbus Behçet".
Su actividad académica fue muy fructífera, publicando durante su vida profesional 137 artículos científicos, 2 libros de textos, 12 monografías y 17 traducciones médicas.1-3
Enfermedad de Behçet
Las primeras referencias sobre esta enfermedad, fueron hechas por Hipócrates alrededor del año 500 antes de Cristo.2
En el año 1930, el oftalmólogo griego Benediktos Adamantiades reportó a un paciente con artritis, úlceras orales y genitales, flebitis, e iritis, pero no fue hasta 1937 que Hulusi Behçet publica su serie de casos que venía estudiando desde 1924, definiéndola como un proceso inflamatorio de etiología ignorada que se identifica por aftas orales recurrentes, úlceras genitales, uveítis y lesiones cutáneas.1,2
En la actualidad se sabe que no resulta una enfermedad contagiosa, ni de trasmisión sexualmente, y que se desarrolla en personas genéticamente propensas, expuestas a algún agente externo medioambiental, y con dificultades en la respuesta inmunológico natural ante organismos infecciosos como virus y bacterias; definiéndola como una vasculitis sistémica que cursa con inflamación de cualquier vaso sanguíneo, en la que se puede afectar prácticamente todos los órganos y sistemas.7
Incidencia y prevalencia
Relacionada inicialmente su distribución con las antiguas vías comerciales, y por su elevada incidencia en los países asiáticos desde el medio este a China, recibió el nombre de enfermedad de la Ruta de Seda, en la actualidad se han descritos casos en todo el mundo.8-10
Afecta principalmente a adultos jóvenes entre la 3da y 4ra década de la vida, aunque se han reportado casos en recién nacidos que remiten espontáneamente a los seis meses de edad.11,12
La mayor incidencia se reporta en Turquía con 420 casos por cada 100 000 habitantes al año. En países como Japón, Corea, China, Irán, y Arabia Saudita fluctúa entre 13 a 22 casos por cada 100 000 habitantes. En los Estados Unidos se reportan de 0.12 a 0.33 casos por cada 100,000 habitantes en los primeros años de vida y la juventud, aumentando a 5.2 casos por cada 100,000 habitantes por encima de los 45 años.13,14
Sin embargo, datos del estudio de residentes de Condado de Olmsted, Minnesota arrojan que por encima de los 45 años la prevalencia aumenta en esta población.14
En cuanto a la raza y el sexo el predominio de la EB es más alto entre el Medio Oriente y poblaciones de Japón.14
Su relación con el sexo varía según el país. En el medio este de Asia, incluyendo Israel, Egipto y Turquía es más común en el sexo masculino, con una proporción entre 3 a 5 varones por cada hembra; los reportes de Alemania, Japón, y Brasil son más frecuentes en hembras que varones, llegando a ser en los Estados Unidos de 5 hembras por cada varón afectado.13-15
En el sexo masculino la enfermedad resulta más grave, con frecuente presencia de aneurismas pulmonares, complicaciones oculares, tromboflebitis y manifestaciones neurológicas, en el sexo femenino es más frecuente encontrar lesiones en piel y mucosas.14,15 Los casos que desarrollan la enfermedad antes de la edad de 25 años son más propensos a desarrollar complicaciones oculares y vasculitis de grande vasos.16-17
Patogénesis
No existe consenso en todo el mecanismo asociado a su patogenia, en lo que más se coincide es que se trata de una enfermedad autoinmune cuyo principalmente órgano de choque son los capilares de diferentes estructuras, produciendo una vasculitis como respuesta inflamatoria que se traduce en la expresión clínica de la enfermedad.18
Entre los rasgos más característicos de su patología se encuentra su asociación con pacientes genéticamente predispuesto por presentar el antígeno mayor de compatibilidad de clase I HLA-B51 y HLA-B5, principalmente en países del medio oriente y así central, que se ha expuesto a algunos de los microrganismos asociados a la enfermedad, como los estreptococos en relación con las ulceras orales o el estafilococo y el propionibacterio asociado a las lesiones dermatológicas.
En este contexto, se desencadena un mecanismo inflamatorio mediado por células naturales Killer y proteínas de shock térmico, capaces de actuar contra células del organismo por mimetismo celular, y dar lugar a las manifestaciones clínicas a través de procesos de inflamación reparación en los vasos sanguíneos de estos tejidos afectados.
El HLA-B51 se ha mostrado para ser más prevaleciente en Turquía, Medio Oriente, y Japón, correspondiendo con un predominio más alto de esta enfermedad dentro de esas poblaciones.19
Rasgos clínicos
Esta entidad se caracterizada por producir una vasculitis en vasos de todos los tamaños, cuyas manifestaciones clínicas más frecuentes son la estomatitis aftosa recurrente, úlceras genitales y uveítis. Además presentan con elevada frecuencia, eritema nodoso, úlceras cutáneas, artritis periféricas, dolores abdominales y melena.
Menos frecuentes pero más graves son las trombosis y aneurismas arteriales, así como las manifestaciones neurológicas, principalmente las que afectan el sistema nervioso central o neuro-Behçet, las cuales son potencialmente mortales. La aftosis oral se presenta como una lesión de bordes bien definidos, rodeada de halo eritematoso y con un fondo amarillento, variables en tamaño y número, suelen ser dolorosas, y afectan habitualmente a cara interna de los labios, mejillas y borde de la lengua. Por lo general regresan en unas dos semanas y curan sin dejar cicatriz.19-20
Las úlceras genitales, de aspecto similar a las orales, por lo general dejan cicatriz cuando curan, y se pueden observar en el examen físico. En los hombres se localizan en el escroto y pene, y en la mujer en labios mayores y menores.21
Las manifestaciones cutáneas más comunes son el eritema nodoso y la pseudofoliculitis, esta última se manifiesta como lesión inicialmente papulosa, sobre la que aparece una pústula estéril que se cubre posteriormente de costra.
Estas lesiones se pueden provocar al pinchar la piel con una guja estéril, dando lugar a la prueba de la patergía característicamente positiva en esta entidad .22-24
Otra forma de manifestarse las lesiones vasculares en la piel es en forma de úlceras cutáneas por vasculitis leucocitoclástica, pustulosa o linfocítica.24
Manifestaciones oculares
La uveítis puede ser la primera manifestación de la enfermedad y preceder a la aparición del proceso sistémico.25,26
La uveítis posterior y la vasculitis retiniana son las manifestaciones oculares más frecuentes. La uveítis anterior con presencia de pus en la cámara anterior del ojo (hipopion), es la manifestación clásica en las descripciones iniciales. La recurrencia y gravedad de las manifestaciones oculares pronostican una evolución no favorable a la ceguera, por lo que se debe tener en cuenta y ser más enérgicos en el tratamiento.27
Manifestaciones vasculares
Las manifestaciones vasculares pueden presentarse como tromboflebitis superficial, trombosis venosa o arteritis. La arteritis se manifiesta por fenómenos oclusivos-trombóticos y aneurismáticos que afectan de forma preferente aorta, arteria pulmonar, arteria poplítea, arteria femoral, arteria subclavia, y menos frecuente carótida común; y son responsables de fenómenos de infartos o hemorrágicos en diferentes órganos.28-30
Estas manifestaciones se producen entre el 15 y el 35 % de los pacientes, predominando en el sexo masculino, y afectan tanto al sistema arterial como al venoso de todos los tamaños y territorios.31-33
Los grandes vasos puede afectarse precozmente, incluso antes de que aparezcan las úlceras orales y sin otras manifestaciones clínicas, presentando dificultades para hacer el diagnóstico.34
El desarrollo de trombosis superficial se asocia con riesgo elevado de presentarse en otras localizaciones y de lesiones arteriales. De éstas 2/3 son aneurismas que pueden observarse en todo el árbol arterial incluidas las coronarias y 1/3 son oclusiones. La arteria que se afecta con mayor frecuencia es la aorta seguida de la pulmonar, femoral, poplítea, subclavias y carótidas.34
La combinación de tromboflebitis con aneurismas arteriales pulmonares, síndrome de Hughes Stovin, se ha sugerido como manifestación vascular única. El retraso en el diagnóstico y en el inicio del tratamiento de esta complicación puede ser responsable de la rotura del aneurisma con hemoptisis masiva de una elevada mortalidad. La base patológica es una vasculitis de la vasa vasorum.34
La participación de otros órganos es menos frecuente. Se han descrito casos de pericaditis, endocarditis, infartos de miocardio y vasculitis coronaria y aneurismas ventriculares. Las alteraciones pulmonares, aneurismas arteriales, trombosis venosa y arterial, infartos pulmonares, también están relacionadas con vasculitis. Rara vez se ve derrame pleural. La TAC puede ayudar al diagnóstico de los aneurismas. Como en otros territorios los estudios histológicos de las lesiones arteriales muestran arteritis de la vasa vasorum. También se han descrito alteraciones renales que van desde glomerulonefritis difusa y focal sin alteración de la función renal a proteinuria y hematuria microscópica.34
Las manifestaciones articulares, presentes en más del 50 % de los pacientes, suelen presentarse como monoartritis o poliartritis, simétricas y no erosivas. Las articulaciones más frecuentemente afectadas son rodillas, tobillos, codos y muñecas.35
Manifestaciones neurológicas
Entre el 10 % y el 20 % de los casos presentan manifestaciones neurológicas; estas suelen estar relacionadas con inflamatoria del sistema nervioso central, y menos frecuente, por afectación vascular a nivel local, o polineuropatía periférica.36,37
Las formas más frecuentes de presentación son la meningoencefalitis asépticas. Otras manifestaciones como los déficits neurológicos, suelen expresarse en forma de trastornos sensitivos, síndrome piramidal, convulsiones, síndrome cerebeloso, síndrome vestibular y parálisis de nervios oculomotores. El compromiso del sistema nervioso central está dado por la vasculitis de vasos intracerebrales (neuro-Behçet), y se corresponde con accidentes vasculares encefálicos o trombosis de pequeños vasos cerebrales o grandes senos venosos que producen hipertensión endocraneal, representando siempre manifestaciones graves de esta enfermedad.38,39
Se han descrito además manifestaciones psiquiátricas expresadas por cuadros confusionales o demencias en periodos avanzados de la enfermedad, así como alteraciones de sistema nervioso periférico (SNP), caracterizadas por polineuritís sensitivo motora distal de las cuatro extremidades y mononeuritis múltiple.40,41
Manifestaciones gastrointestinales
Las manifestaciones gastrointestinales suelen presentarse en forma de dolor abdominal, diarreas y melenas; con menor frecuencia pero mayor gravedad puede aparecer úlceras ileocecales largas y profundas en el área opuesta al mesenterio (rasgo característico de esta enfermedad) que facilitan la perforación intestinal. La región ileocecal es la más afectada, seguida por colon transverso, ascendente y esófago.42,43

Diagnóstico diferencial
El diagnóstico diferencial suele estar claro en la mayoría de los casos, sin embargo hay pacientes con cuadros abigarrados que se sitúan entre la enfermedad inflamatoria intestinal crónica y la EB. Estas dos entidades comparten además gran número de manifestaciones extraintestinales como las úlceras orales, el eritema nodoso, la uveítis y la artritis. La histología muchas veces tampoco permite diferenciar ambas entidades. Suele utilizarse la presencia de granulomas en las úlceras intestinales como diagnóstico de enfermedad de Crohn y la presencia de una prueba de patergia cutánea positiva como signo de EB.44
Baixauli en su original aporta una tabla que ofrecemos adaptada como ayuda para el diagnóstico diferencial de los síntomas y las úlceras en la enfermedad de Behçet. [Tabla 2]
Son las ulceraciones en mucosas, orales y genitales el primer criterio diagnóstico, estas suelen aparecer como la primera manifestación y la única durante años.45
Aunque se les han atribuido ciertas características a estas lesiones lo cierto es que son indistinguibles de muchas lesiones ulcerosas y la histología es inespecífica con evidencia de vasculitis.46
En los períodos iniciales debe considerarse el diagnóstico diferencial con herpes simple.47
Otras lesiones cutáneas frecuentes pueden adquirir diversas formas: nodulares semejantes a las del eritema nudoso con rangos histopatológicos de reacción vascular neutrofílica, púrpura palpable, vasculitis necrotizante y pápulas y pústulas como las del acné vulgar o foliculitis. Puesto que estas últimas lesiones son inespecíficas, particularmente en adolescentes, algunos autores consideran que no deberían incluirse entre los criterios diagnósticos.48
En el aparato digestivo la ulceración del tracto gastrointestinal es la lesión característica y la localización más frecuente la mucosa del íleon terminal y ciego y, en menor grado, de otros territorios del colon. También puede afectarse el esófago. El diagnóstico diferencial con otras enfermedades intestinales, en particular con la colitis ulcerosa, no es fácil. Los cambios histológicos son similares y las manifestaciones extraarticulares, como ulceras orales, eritema nudoso, uveítis y artritis, indistinguibles por lo que es preciso excluir una enfermedad inflamatoria intestinal antes de establecer el diagnóstico de EB.49
La mitad de los pacientes con EB tiene artritis, oligo o monoarticular, no erosiva que afecta a las grandes articulaciones de los miembros inferiores. La artritis no se limita a las articulaciones periféricas. Además se encuentra sacroileítis, en general sin dolor lumbar inflamatorio.50
Aquellos que son tratados en forma precoz presentan secuelas menos severas que los pacientes que reciben tratamiento tardío, con secuelas graves, imposibilitan su reinserción laboral o reincorporación a estudios superiores.
Ha sido reportado que la ciclosporina A podría inducir manifestaciones neurológicas en pacientes con EB.51
Puede presentar Síndrome Pulmón-Riñón (SPR por el mecanismo de la lesión a través del depósito de inmunocomplejos (IgG, C3 y C4) con capilaritis pulmonar y glomerulonefritis (GN).52,53
Diagnóstico
La EB no tiene manifestaciones clínicas patognomónicas, ni exámenes de laboratorio específicos, por lo que el diagnóstico se realiza en base a criterios clínicos predefinidos según distintos grupos de estudio. Dentro de éstos, los más utilizados son los de Behget's Disease Research Committee of Japan (BDCJ), y los del International Study Group for Behget's Disease (ISG).5,6
La mayoría de los pacientes pueden clasificarse de acuerdo a los criterios del ISG creados en 1990, los cuales poseen una excelente especificidad, pero carecen de sensibilidad, los que fueron modificados en el 2006 por el grupo internacional para el estudio de la EB. En esa última revisión fueron incluidos las lesiones vasculares (trombosis arteriales, venosas, aneurismas) como criterio diagnóstico, además de los criterios ya conocidos, descritos en la tabla 1.
No obstante la existencia de formas incompletas de la enfermedad y de diferencias regionales en la prevalencia de algunas de las manifestaciones clínicas características puede hacer que el diagnóstico se retrase años o no llegue a establecerse.
El test de patergia incluido entre los criterios de diagnóstico tiene una prevalencia, elevada entre los pacientes del Extremo Oriente, sin embargo es poco frecuente en los del área mediterránea y América y por lo tanto con poca sensibilidad en estas regiones entre las nos incluimos. Su positividad se asocia con un riesgo aumentado de enfermedad vascular.54
No es exclusivo de la EB ya que puede ser positivo en el síndrome de Sweet y en el pioderma gangrenoso. El mecanismo patogénico de esta reacción no es bien conocido y tampoco es exclusivo de la piel. Son varios los tejidos que muestran hiperreactividad a los traumatismos y no es infrecuente la aparición de uveítis tras cirugía ocular o de una sinovitis a continuación de una artrocentesis.54
La inflamación ocular, con una prevalencia entre el 47 y 75 %, consiste en una uveítis recidivante, en general bilateral, que puede afectar tanto a la cámara anterior como a la posterior o a ambas. Suele aparecer después de 2-3 años del comienzo de la enfermedad y en ocasiones es el primer síntoma. En un 20 % de los pacientes puede observarse hipopión que aunque característico no es específico de la EB y puede verse en algunas artropatías asociadas al HLA-B27. La lesión retiniana, la más severa, es una vasculitis con producción de hemorragias y trombosis venosas. Es casi constante el edema macular quístico y la papilitis. Después de cada recidiva queda alguna lesión estructural aunque en los últimos años con el reconocimiento precoz de la enfermedad y los nuevos tratamientos el pronóstico ha mejorado considerablemente. Algunas retinitis virales y la sarcoidosis pueden producir lesiones indistinguibles de las de la EB.55
Los criterios diagnósticos actuales para el Behçet permitieron diagnosticar un mayor número de pacientes que los ISG a Wurmann y col. 2009. Aunque los criterios BDCJ son más exigentes que los ISG para el diagnóstico de EB completa (pues requiere de 4 síntomas típicos), ambos criterios son similares para el diagnóstico de esta enfermedad en su modalidad incompleta. Sin embargo, los criterios ISG dan una mayor importancia a la presencia de úlceras orales (ya que en su ausencia no se puede diagnosticar la enfermedad) mientras que los BDCJ otorgan más relevancia al compromiso ocular. Esta diferencia se puede explicar por el hecho que esta entidad tiene variabilidad en su expresión clínica en las distintas etnias. Por ejemplo la EB es una de las principales causas de amaurosis en Japón y Turquía, lo que no ocurre en Europa. Otra diferencia es que el test de patergia está incluido en los criterios ISG y no en los BDCJ. En este punto debemos considerar que la aplicación clínica del test de patergia no fue frecuente en Chile y cuando fue realizado en general resultó negativo. Por lo anteriormente expuesto los criterios BDCJ podrían contribuir a disminuir el sub diagnóstico de EB en nuestro medio.5,6,56
Exámenes complementarios
Leucocitosis a predominio de polimorfonucleares, los reactantes de la fase aguda suelen ser anormales durante las crisis, principalmente la eritrosedimentación que suele estar muy acelerada, transaminasa glutámico oxalacetica aumentada, ultrasonido abdominal hígado reactivo, resto de estructuras anatómicas normales (vesícula, páncreas, bazo y riñones) en tamaño y forma, no adenopatías paravertebrales, tomografía axial computarizada de tórax no se observan alteraciones en los cortes realizados, Fondo de ojos no alteraciones vasculares o retinianas, Exudado vaginal presencia de células de descamación sucia, no trichomonas ni levaduras.57,58
Los estudios histopatológicos han sido realizados principalmente en pústulas cutáneas y en ellos se observa mayoritariamente una reacción vascular neutrofílica.59
Estudios con inmunofluorescencia indirecta en biopsias de pulmón y riñón en pacientes con EB con capilaritis pulmonar y glomerulonefritis revelan depósitos de inmunocomplejos granulares en paredes de vénulas y capilares de IgG, C3, C4 y fibrinógeno.60
Las alteraciones del líquido cefalorraquídeo son inespecíficas, con pleocitosis de polimorfonucleares y linfocitos.61En la resonancia magnética pueden observarse imágenes focales de hiperseñal en T2, en el tronco cerebral, ganglios basales y sustancia blanca. Estas imágenes no son específicas de la EB y aparecen en otras enfermedades que cursan con vasculitis.62
La causa de la tendencia a la trombosis en la EB no es bien conocida y tampoco la importancia de las alteraciones de la coagulación observadas. La elevación de los niveles de antitrombina III que muestran algunos pacientes puede deberse a mecanismos compensatorios contra el incremento de la actividad procoagulante en pacientes con tromboembolismo. La tomografía axial computarizada puede ayudar al diagnóstico de los aneurismas.63
En la biopsia de las lesiones ulcerativas suele encontrarse infiltrado leucocitario en dermis superficial, necrosis epidérmica y presencia de leucocitos polimorfonucleares sin lesión tumoral.63
Aproximadamente en el 50 % de los casos se demuestra la presencia de anticuerpos circulantes contra la membrana mucosa de la cavidad bucal.64
Etiología
La EB también puede aparecer en la infancia, en general con formas incompletas. Las manifestaciones más comunes son las úlceras orales y genitales, lesiones cutáneas, artritis y uveítis. Algunos recién nacidos de mujeres con EB presentan úlceras orales y genitales, pústulas y úlceras necróticas cutáneas de distribución periungueal. Estas lesiones son autolimitadas y desaparecen en seis semanas. El curso de la RB durante el embarazo es variable. Puede producirse activación, remisión o cambios clínicos en cada caso.65,66
Gül en un estudio con 16 mujeres con EB durante la gestación y posterior parto, observó que las manifestaciones clínicas de actividad durante el embarazo consisten en lesiones mucocutáneas y artritis. Inflamaciones oculares y tromboflebitis se produjeron en pocas ocasiones. Todos los embarazos llegaron a término y los niños nacieron sanos. Las reactivaciones post-parto fueron escasas.66
Otras enfermedades que pueden presentar lesiones inflamatorias de las mismas características a las observadas en la EB son la enfermedad de Crohn, la colitis isquémica de predominio derecho, la tuberculosis intestinal y la infección por Yersinia spp., entre otras.67
Se han sugerido como agentes causales a bacterias y virus, aunque no se posee ninguna prueba convincente.67
La exposición a un agente infeccioso puede activar una respuesta inmune cruzada- reactiva. Los agentes infecciosos propuestos han incluido el virus de herpes simple (HSV), especies del Estreptococo, especies de Estafilococo, y Escherichia coli; de los cuales todos normalmente habitan la cavidad oral.67
El estudio de proteínas de choque térmico como human HSP-60 y HSP-65 ha proporcionado alguna visión en posibles mecanismos que contribuyen al desarrollo de la EB. Estas se encuentren en concentraciones altas en las úlceras orales y las lesiones superficiales activas.68,69
Anatomía Patológica y Fisiopatológica
La prevalencia de HLA B5 entre los pacientes es alta y más específicamente la del alelo B51 que se considera un importante factor de riesgo en áreas en que la incidencia de Enfermedad de Behçet es mayor como sucede en los países asiáticos.70
Entre los factores ambientales algunas infecciones parecen implicadas en el desarrollo de la enfermedad. Se ha aislado DNA del virus del herpes simple en núcleos de linfocitos de sangre periférica y en el suero se han detectado anticuerpos contra el virus.71
También se ha observado una elevada incidencia de amigdalitis y caries dentales causadas por estreptococos y el estudio histológico de las ulceraciones orales revelaba infiltrados inflamatorios de neutrófilos, linfocitos T y depósitos de componentes de estreptococos.72
La alteración celular característica de la EB es el incremento de migración de los neutrófilos, células predominantes en las lesiones iniciales. El principal hallazgo microscópico de las lesiones activas de la EB es una vasculitis oclusiva mediada por mecanismos inmunes. El infiltrado perivascular revela infiltrados de linfocitos T, CD4+ y CD8+ y células HLA-DR.73
Tratamiento, evolución, pronóstico y rehabilitación
El tratamiento, con frecuencia poco satisfactorio, es sintomático y depende de las manifestaciones clínicas. A la hora de planificarlo hay que tener en cuenta que la enfermedad cursa con remisiones y exacerbaciones impredecibles, que el comienzo a edad temprana y el sexo masculino son factores de mal pronóstico. La participación de órganos vitales marca la toma de decisiones.74
Las prioridades son las complicaciones gastrointestinales, las del sistema nervioso central y las de los grandes vasos que con frecuencia requieren dosis elevadas de corticoides e inmunosupresores y en ocasiones intervenciones quirúrgicas.74,75
La lesiones mucocutáneas responden en general al tratamiento con corticoides tópicos o intralesionales. La colchicina, usada en el tratamiento de las lesiones mucocutáneas de la EB desde el año 1975, resulta útil debido a su acción inhibitoria sobre la función de los neutrófilos.
La talidomida, que a dosis de 100 mg/día muestra la misma efectividad que 300 mg/día, ha demostrado ser muy eficaz en la resolución de las lesiones mucocutáneas refractarias a otros tratamientos. Su mecanismo de acción es a través de la inhibición del TNF-α y de la angiogénesis.76
Su mayor inconveniente es la teratogenicidad y la neuropatía periférica que produce. La dapsona oral a dosis entre 50 y 100 mg/día también ha resultado ser útil en pacientes con manifestaciones mucocutáneas persistentes aunque su uso en la EB es muy restringido.76
En casos de enfermedad mucocutánea grave están indicados los corticoides sistémicos. El tratamiento de las lesiones oculares depende de su severidad.76
Comienzo temprano y sexo masculino son factores de riesgo de complicaciones oculares severas. Los agentes citotóxicos o inmunosupresores como azatioprina, clorambucilo, ciclofosfamida pueden prevenir la aparición de nuevos episodios en el 50 % de los pacientes.77
La ciclofosfamida es la más eficaz en la vasculitis retiniana. En pacientes refractarios a otros tratamientos la ciclosporina sola o en combinación con azatioprina puede ser eficaz.
Este tratamiento está contraindicado en pacientes con síntomas neurológicos y con lesiones neurálgicas subclínicas detectadas por resonancia magnética porque puede ser neurotóxica en pacientes con EB.
Recientes estudios han mostrado esperanzadores resultados con interferón-alfa aunque no se conoce la dosis adecuada y se produce una reactivación al suspender el tratamiento.78
El interferón alfa también produce una marcada reducción de las lesiones mucocutáneas y de la vasculitis. De la misma forma, los anticuerpos anti-TNF-alfa han demostrado ser muy eficaces en la supresión de la uveítis posterior refractaria, y otras afecciones relacionadas con la vasculitis.79
El tratamiento en los pacientes con oclusiones venosas continúa siendo controvertido. Va desde el uso de antiagregantes plaquetarios hasta anticoagulación oral o parenteral.80
Los pacientes con vasculitis y aneurismas pueden beneficiarse de la terapia con ciclofosfamida oral o en pulsos mensuales, sola o en combinación con esteroides sistémicos.80
Las lesiones gastrointestinales requieren sulfasalacina y esteroides. La cirugía se considerará en los pacientes con perforación intestinal y sangrado persistente.79,80
Para prevenir complicaciones postquirúrgicas por la reacción inflamatoria local característica en la EB, los pacientes deben recibir esteroides durante varios días.79,80
El tratamiento de las fases agudas de la enfermedad neurológica se basa en altas dosis de esteroides, orales o en pulsos intravenosos, seguidos de dosis medias orales durante varios meses. Pueden asociarse ciclofosfamida o metotrexato. La enfermedad progresiva crónica del sistema nervioso central es en general resistente a las terapias disponibles hoy.79,80
El 25 % de los que tienen lesiones oculares pueden perder la visión.79,80
Las causas de muerte siguen siendo la ruptura de aneurismas, la enfermedad severa del SNC y la perforación intestinal por lo que es imprescindible identificar precozmente estas complicaciones para aplicar medidas terapéuticas adecuadas.
Sorprendentemente algunos pacientes con trombosis masiva de la cava pueden sobrevivir durante años al desarrollar una extensa red colateral de venas superficiales y varices.79,80
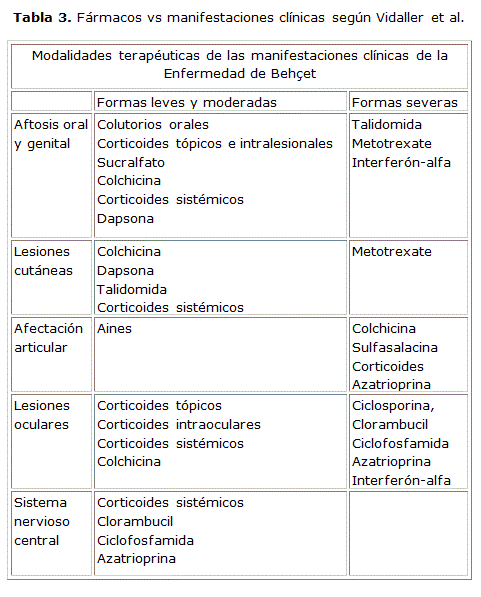
Vidaller et propone para cada manifestación clínica una serie de fármacos [Tabla 3]81
Rehabilitación
La EB es una enfermedad crónica que puede desaparecer y reaparecer independientemente del tratamiento. Esto a veces hace que el análisis de los beneficios de la terapia sea difícil de hacer.
La mayoría de los pacientes con EB viven una vida intensa, aunque lo más seguro es que tengan que lidiar con algunos de los síntomas en algún nivel durante su vida.
La muerte ocurre en alrededor del 4% de los casos de EB. Las causas de la muerte suelen ser más a menudo perforación intestinal; derrames cerebrales; y rotura de vasos sanguíneos que estaban engrandecidos y debilitados como sucede en los aneurismas.82,83
La evolución de la EB suele ser intermitente, con períodos de remisión (falta de actividad de la enfermedad) y de exacerbación (períodos de actividad) a lo largo de los años, con una tendencia progresiva hacia la remisión. Los síntomas pueden durar desde días a semanas, o pueden persistir durante meses o años. Suelen provocar discapacidad que disminuye la calidad de vida.84
CONCLUSIONES
La EB es una enfermedad inflamatoria crónica, recurrente, y multisistémica de causa desconocida caracterizada por úlceras orales recurrentes, úlceras genitales, uveítis y lesiones cutáneas. Es más frecuente entre el este asiático y el Mediterráneo, por lo que también se la conoce como enfermedad de la ruta de la seda. Todas las manifestaciones son autolimitadas excepto la afección ocular. La afección del tracto gastrointestinal, sistema nervioso central y grandes vasos es menos frecuente, aun así pueden amenazar la vida del paciente. La susceptibilidad de la EB está asociada con la presencia del alelo HLA-B51.
REFERENCIAS BIBLIOGRÁFICAS
1. Collazo Borrego L, Soto Escobar A, Averhoff Casamayor MC, Turcaz Bosh N, Ramos Sánchez V, Torres Escalonas N. Síndrome de Behcet. Reporte del primer caso en Cuba. Rev Cubana Estomatol. 1989;26(3):175-80.
2. Mutlu S, Scully C. The person behind the eponym: Hulûsi Behçet (1889-1948). J Oral Pathol Med. 1994;23(7):289-96.
3. Tunc R, Uluhan, A, Melikoglu M, Ozyazgan Y, Ozdogan H, Yazici H. A reassessment of the International Study Group criteria for the diagnosis (classification) of Behçet's syndrome. Clinical and experimental rheumatology. 2001;19(5 Suppl 24):S45.
4. Yazici H, Yazici Y. Behçet's syndrome. London: Ed. Springer; 2010. p. 231-38.
5. O'Neill, TW, Rigby AS, Silman AJ, Barnes C. Validation of the International Study Group criteria for Behçet's disease. Rheumatology. 1994;33(2):115-7.
6. International Team for the Revisin of the Internacional Criteria for Behçet's Disease (ITR-ICBD). Revision of the International Criteria for Behçet's Disease (ICBD). Clin Exp Rheumatol. 2006;24 Suppl 42:S14-5.
7. Ginarte M, Toribio J. Síndrome de Sweet. Medicina clínica. 2009;133(1):31-5.
8. Khairallah M, Accorinti M, Muccioli C, Kahloun R, Kempen J. Epidemiology of Behcets disease. Ocul Immunol Inflamm. 2012;20(5):324-35.
9. Soto-Rojas AE, Kraus A. The oral side of Sjögren syndrome. Diagnosis and treatment. A review. Arch Med Res. 2002;33(2):95-106.
10. Carreño Pérez L. Enfermedad de Behçet. An. Med. Interna [revista en Internet]. 2001 Ago [citado 18 abril 2014];18(8):5-10. Disponible en: http://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S0212-71992001000800001&lng=es
11. Hatemi G, Bahar H, Uysal S, Mat C, Gogus F, Masatlioglu S, Yazici H. The pustular skin lesions in Behcet’s syndrome are not sterile. Annals of the rheumatic diseases. 2004;63(11):1450-2.
12. Ozen S. Familial Mediterranean fever: revisiting an ancient disease. European journal of pediatrics. 2003;162(7-8):449-54.
13. Davatchi F, Shahram F, Chams-Davatchi C, Shams H, Nadji A, Akhlaghi M, Abdollahi BS. Behcet’s disease: from East to West. Clinical rheumatology. 2010;29(8):823-33.
14. Kitaichi N, Miyazaki A, Iwata D, Ohno S, Stanford MR, Chams,H. Ocular features of Behçet’s disease: an international collaborative study. British Journal of Ophthalmology. 2007;91(12):1579-82.
15. López de Maturana LD, Amaro BP, Balestrini DC, Segovia GL. Manifestaciones clínicas en 5 casos de enfermedad de Behçet: Report of 5 cases. Rev. méd. Chile [revista en Internet]. 2002 Mayo [citado 12 mayo 2014];130(5):551-56. Disponible en: http://www.scielo.cl/scielo.php?script=sci_arttext&pid=S0034-98872002000500010&lng=es&nrm=iso&tlng=es
16. Hernández GAM, Sanabria CR.). Enfermedad de Behcet. Revista medica de costa rica y Centroamérica. 2014;71(611):523-8.
17. Mendes D, Correia M, Barbedo M, Vaio T, Mota M, Gonçalves, O, Valente J. Behçet's disease a contemporary review. Journal of autoimmunity. 2009;32(3):178-88.
18. Yazici H, Fresko I, Yurdakul S. Behçet's syndrome: disease manifestations, management, and advances in treatment. Nature Clinical Practice Rheumatology. 2007;3(3):148-55.
19. Seyahi E, Melikoglu M, Yazici H. Clinical features and diagnosis of Behcet's syndrome. Int J Adv Rheumatol. 2007;5(1):8-11.
20. Mat C, Yurdakul S, Uysal S, Gogus F, Ozyazgan Y, Uysal O, Yazici H. A double-blind trial of depot corticosteroids in Behcet's syndrome. Rheumatology. 2006;45(3):348-52.
21. Lees CW, Barrett JC, Parkes M, Satsangi J. New IBD genetics: com pathways with other diseases. Gut. 2011;60(12):1739-53.
22. Saadoun D, Wechsler B, Resche‐Rigon M, Trad S, Le Thi Huong D, Sbai A, Piette JC. Cerebral venous thrombosis in Behçet's disease. Arthritis Care & Research. 2009;61(4):518-26.
23. Alpsoy E, Zouboulis C, Ehrlich GE. Mucocutaneous lesions of Behçet's disease. Yonsei medical journal. 2007;48(4),573-85.
24. Yi SW, Cheon JH, Kim JH, Lee SK, Kim TI, Lee YC, Kim WH. The prevalence and clinical characteristics of esophageal involvement in patients with Behçet's disease: a single center experience in Korea. Journal of Korean medical science. 2009;24(1):52-6.
25. Tugal-Tutkun I, Onal S, Altan-Yaycioglu R, Huseyin Altunbas H, Urgancioglu M. Uveitis in Behçet disease: an analysis of 880 patients. Am J Ophthalmol. 2004;138(3):373-78.
26. Gül A, Tugal-Tutkun I, Dinarello CA, Reznikov L, Esen BA, Mirza A, Solinger A. Interleukin-1β-regulating antibody XOMA 052 (gevokizumab) in the treatment of acute exacerbations of resistant uveitis of Behçet's disease: an open-label pilot study. Annals of the rheumatic diseases. 2012;71(4):563-6.
27. Bañares A, Pato E, Abasolo L. Treatment of refractory posterior uveítis with anti-TNF alfa (Infliximab). Ann Rheum. Dis. 2001;60 Suppl 1:S145-9.
28. Sarica-Kucukoglu R, Akdag-Kose A, Kayabal IM, Yazganoglu KD, Disci R, Erzengin D, Azizlerli G. Vascular involvement in Behçet's disease: a retrospective analysis of 2319 cases. Int J Dermatol. 2006;45(8):919-21.
29. Paller AS, Mancini AJ. Hurwitz clinical pediatric dermatology: a textbook of skin disorders of childhood and adolescence. Chapter 25. New York: Ed. Elsevier Health Sciences; 2011.
30. Seyahi E, Melikoglu M, Akman C, Hamuryudan V, Ozer H, Hatemi G, Yazici H. Pulmonary artery involvement and associated lung disease in Behçet disease: a series of 47 patients. Medicine. 2012;91(1):35-48.
31. Caramaschi P, Poli G, Bonora A, Volpe A, Tinazzi I, Pieropan S, Biasi D. A study on thrombophilic factors in Italian Behcet's patients. Joint Bone Spine. 2010;77(4):330-4.
32. Ramos-Casals M, Diaz C, Cuadrado MJ, Khamashta, MA. Autoimmune diseases induced by biological agents: a double-edged sword? Autoimmunity reviews. 2010;9(3):188-93.
33. Branch DW, EllerAG. Antiphospholipid syndrome and thrombosis. Clinical obstetrics and gynecology. 2006;49(4):861-74.
34. Tabbara KF, Al-Hemidan AI. Infliximab effects compared to conventional therapy in the management of retinal vasculitis in Behçet disease. American journal of ophthalmology. 2008;146(6):845-50.
35. Saadoun D, Bodaghi B, Bienvenu B, Wechsler B, Sene D, Trad S, Sève P. Biotherapies in inflammatory ocular disorders: interferons, immunoglobulins, monoclonal antibodies. Autoimmunity reviews. 2013;12(7):774-83.
36. Al-Araji A, & Kidd DP. Neuro-Behçet's disease: epidemiology, clinical characteristics, and management. The Lancet Neurology. 2009;8(2):192-204.
37. Salas Cabrera R, Sagué Larrea J, Laurencio Mena A. Enfermedad de Behcet: Presentación de un caso. Arch. Esp. Urol. [revista en Internet]. 2007 Feb [citado 19 noviembre 2013];60(1):67-8. Disponible en: http://scielo.isciii.es/pdf/urol/v60n1/caso1.pdf
38. Sumida T, Tsuboi H, Iizuka M, Nakamura Y, Matsumoto I. Functional role of M3 muscarinic acetylcholine receptor (M3R) reactive T cells and anti-M3R autoantibodies in patients with Sjögren's syndrome. Autoimm. reviews. 2010;9(9):615-7.
39. Gotkine M, Vaknin-Dembinsky A. Central nervous system vasculitis. Current treatment options in neurology. 2013;15(3):367-74.
40. Hatemi G , Silman A, Bang J, Bodaghi B, Chamberlain AM, Gul A, Yazici H. EULAR recommendations for the management of Behçet disease. Annals of the rheumatic diseases. 2008;67(12):1656-62.
41. Arida A, Fragiadaki K, Giavri E, Sfikakis PP. Anti-TNF agents for Behcet's disease: analysis of published data on 369 patients. In Seminars in arthritis and rheumatism. 2011;(41(1):61-70.
42. Mathews SA, Kurien BT, Scofield RH. Oral manifestations of Sjögren's syndrome. J Dent Res. 2008;87(4):308-18.
43. Voulgarelis M, Tzioufas AG. Pathogenetic mechanisms in the initiation and perpetuation of Sjögren's syndrome. Nat Rev Rheumatol. 2010;6(9):529-37.
44. Ebert EC. Gastrointestinal manifestations of Behçet’s disease. Digestive diseases and sciences. 2009;54(2):201-07.
45. Millán Parrilla F, Quecedo Estébanez E, Gimeno Carpio E. Tratamiento de la estomatitis aftosa recidivante. Piel. 2010;25(8):463-9.
46. Mutinga ML, Odze RD, Wang H H, Hornick JL, Farraye, FA. The clinical significance of right‐sided colonic inflammation in patients with left‐sided chronic ulcerative colitis. Inflammatory bowel diseases. 2004;10(3):215-9.
47. Kötter I, Günaydin I, Zierhut M, Stübiger N. The use of interferon α in Behçet disease: review of the literature. In Seminars in arthritis and rheumatism. 2004;33(5):320-35.
48. Mizuki N, Meguro A, Tohnai I, Gül A, Ohno S, Mizuki N. Association of major histocompatibility complex class I chain-related gene A and HLA-B alleles with Behçet's disease in Turkey. Japanese journal of ophthalmology. 2007;51(6):431-6.
49. Evereklioglu C. Current concepts in the etiology and treatment of Behçet disease. Survey of ophthalm. 2005;50(4):297-350.
50. Pozo González A, Barbán Fernández L, Rodríguez del Valle KM, Betancourt Fernández I. Síndrome de Behçet: presentación de un caso. AMC [revista en Internet]. 2010 Dic [citado 5 Septiembre 2014];14(6):1-7. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1025-02552010000600016&lng=es
51. Biester S, Deuter C, Michels H, Haefner R, Kuemmerle-Deschner J, Doycheva D, Zierhut M. Adalimumab in the therapy of uveitis in childhood. British Journal of Ophthalmology. 2007;91(3):319-24.
52. Yılmaz S, Cimen KA. Pulmonary artery aneurysms in Behçet’s disease. Rheumatology international. 2010;30(10):1401-3.
53. Martínez Larrarte JP, Cepero Morales R, Sosa Almeida M, Molinero Rodríguez C, Lantigua Martell MA. Síndrome de Behcet: Presentación de un caso. Rev cubana med [revista en Internet]. 1998 Dic [citado 23 marzo 2014];7(4):249-52. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034-75231998000400007&lng=es
54. Toro A, Pinto L, Velásquez C, Márquez J. Enfermedad de Behcet. Revista Colombiana de Reumatología. 2009;16(1):97-111.
55. Jennette1 JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, Flores-Suarez LF, et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis & Rheumatism. 2013;65(1):1-11.
56. Wurmann Pamela, Díaz Gonzalo, Sabugo Francisca, Soto Lilian, Solanes Federica, Pino Sandra et al. Retrospective review of 44: Chilean patients with Behçet disease. Rev. méd. Chile [revista en Internet]. 2009 Oct [citado 23 marzo 2014];137(10):1333-40. Disponible en: http://www.scielo.cl/scielo.php?pid=S0034-98872009001000008&script=sci_arttext&tlng=en
57. Ozkan F, Cetin GY, Bakan B, Kalender A, Yuksel M, Ekerbicer HC, Sayarlioglu M. Sonographic Evaluation of Subclinical Entheseal Involvement in Patients With Behçet Disease. American Journal of Roentgenology. 2012;199(6):723-9.
58. Bayetto K, Logan RM. Sjögren's syndrome: a review of aetiology, pathogenesis, diagnosis and management. Aust Dent J. 2010;55 Suppl 1:39-47.
59. Beers Mark H, Porter Robert S. Manual de Merck. En: Schumacher Ralph H, Enfermedades difusas del tejido conjuntivo: Síndrome de Behcet. Madrid: Ed. Elsevier; 2006. p. 294-6.
60. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario, F, Watts RA. 2012 revised international chapel hill consensus conference nomenclature of vasculitides. Arthritis & Rheumatism. 2013;65(1):1-11.
61. Fonseca A, Rocha-Filho P, Melo A. Neuro-Behçet: differential diagnosis of recurrent meningitis. Rev Med Chile. 2013;141:114-8.
62. Saldarriaga-Rivera L, da-Silveira-Campos G, Delgado-Quiroz L, Rios-Gomes-Bica B. Síndrome de la vena cava en paciente con Enfermedad de Behçet. Revista Cubana de Reumatología [revista en Internet]. 2014 [citado 15 julio 2014];16(2):[aprox. 4 p.]. Disponible en: http://www.revreumatologia.sld.cu/index.php/reumatologia/article/view/283
63. Rodés Teixidor J, Guardia Massó J. Tratado de Medicina Interna. En: Vázquez Rodríguez JJ, Peña Sánchez de Rivera JM, editores. Otras 9 Enf. inflamatorias sistémicas de etiología desconocida. Barcelona: Ed. Masson SA; 2006. p. 3250-2
64. Leiba L, Seligsohn V, Sidi Y, Harats D, Sela B, Griffin J, et al. Thrombophilic factors in not leading cause of thrombosis of Behcet’s disease. Ann Rheum Dis. 2004;63:1445-9.
65. Hwang I, Lee CK, Yoo B, Lee I. Necrotizing villitis and decidual vasculitis in the placentas of mothers with Behçet disease. Human pathology. 2009;40(1):135-38.
66. Jadaon J, Shushan A, Ezra Y, Sela H Y, Ozcan C, Rojansky N. Behçet's disease and pregnancy. Acta obstetricia et gynecologica Scandinavica. 2005;84(10):939-44.
67. Grigg E, Kane S, Katz S. Mimicry and Deception in Inflammatory Bowel Disease and Intestinal Behçet Disease. Gastroenterology & Hepatology. 2012;8(2):103-9.
68. Direskeneli H. Autoimmunity vs autoinflammation in Behcet's disease: do we oversimplify a complex disorder? Rheumatology. 2006;45(12):1461-5.
69. Birtas-Atesoglu E, Inanc N, Yavuz S, Ergun T, Direskeneli H. Serum levels of free heat shock protein 70 and anti-HSP70 are elevated in Behcet's disease. Clinical & Experimental Rheumatology. 2008;26(4):S96.
70. Mora Hernández GA, Ríos Sanabria C. Enfermedad de Behcet. Revista medica de costa rica y Centroamérica. 2014;LXXI(611):523-8.
71. Pila Pérez RV, Pila Peláez RU, Rosales Torres P, Artola González JA. Enfermedad de Behcet: presentación de un caso. AMC [revista en Internet]. 2014 Feb [citado 18 julio 2014];18(1):134-45. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1025-02552014000100014&lng=es
72. Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nature Reviews Immunology. 2006;6(11):823-35.
73. Mahammad A, Mandl T, Stufeet C, Segel Mark M. Incidence, prevalence and clinical characteristics of Behcet’s disease in southern Sweden. Rheumatology(Oxford). 2013;52(2):304-10.
74. García-Palenzuelaa R, Graña J, Varela M, Tovar M. Actualización de la enfermedad de Behcet. A propósito de 2 casos en atención primaria. Semergen. 2012;38(1):33-9.
75. Álvarez Álvarez G, Martínez Delgado F, González Cortiñas Mo, Martín García CL, Martínez Fraga A. Enfermedad de Behcet: Informe de 5 casos. Rev cubana med [revista en Internet]. 1998 Jun [citado 7 marzo 2014];37(2):119-22. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034-75231998000200009&lng=es
76. Vandaele P, Kappen J, Van Hagen P, Van Laar J. Managing Behcet’s disease, an update on current and emerging therapies. Ther Clin Res Manag. 2009;5:385-90.
77. Cruz Santana AN, Antunes T, Monteiro de Barros J, Adib Kairalla R, Ribeiro de Carvalho CR, Valente Barbas CS. Acometimento pulmonar na doença de Behçet: uma boa experiência com o uso de imunossupressores. J bras pneumol [serie en Internet]. 2008 [citado 18 abril 2014];34(6):[aprox. 9 p.]. Disponible en: http://www.scielo.br/scielo.php?pid=S1806-37132008000600005&script=sci_arttext&tlng=pt
78. Marques Zaghetto JP, Mina Yamamoto M, Barreto Souza M, Bezerra Gaspar Carvalho da Silva FT, Hirata CE, Olivalves E, et al. Chlorambucil and cyclosporine A in Brazilian patients with Behçet's disease uveitis – a retrospective study. Arq Bras Oftalmol [serie en Internet] 2010 [citado 18 abril 2014]; 73(1):[aprox. 11 p.]. Disponible en: http://www.scielo.br/scielo.php?pid=S0004-27492010000100007&script=sci_arttext&tlng=es
79. Sfikakis P, Markomichelakis N, Alpsoy E, Assad Khalil S, Bodaghi B, Gul A, et al. Anti-TNF therapy in the management of Behcet is disease: Review and basis recommendations. Rheumatology. 2007;46(5):136-41.
80. Santos A. Botequio L, Ribeiro E, Botega J. Alterações psiquiátricas após corticoterapia em paciente com rara manifestação neurológica de Síndrome de Behçet e o papel da interconsulta psiquiátrica. Rev Psiq Clín. 2009;36(5):203-5.
81. Vidaller Palacín A, Robert Olalla J, Sanuy Jiménez B, Rufi Rigau G, Folch Civit J, Charte González A. Tratamiento de la enfermedad de Behçet. An. Med. Interna. 2002;19(11):52-6.
82. Karakaya K, Comert M, Numanoglu G. Multiple perforations along the transverse colon as a rare presentation of intestinal behcet's disease: a case report. Clinics [serie en Internet]. 2009 [citado 12 mayo 2014];64(12):[aprox. 6 p.]. Disponible en: http://www.scielo.br/scielo.php?pid=S1807-59322009001200016&script=sci_arttext
83. Balaguer J, Tarancón B, Novo E, García S. Afectación pericárdica en enfermedades de patogenia inmunológica, infecciosa y endocrinológicas. Medicine-Programa de Formación Médica Continuada Acreditado. 2013;11(43):2583-90.
84. Harrison Online [homepage on the internet]. EEUU: Síndrome de Behçet; c1995-2010[actualizado 9 abril 2010; [citado 18 de abril de 2014]. Harrisonmedicina [aprox. 2 pantallas]. Disponible en: http://www.harrisonmedicina.com/content.aspx?aID=3743565&searchStr=s%c3%adSindrome+de+beh%c3%a7et#3743565
Los autores refieren no tener conflicto de intereses.
Recibido: 18 de febrero de 2014
Aprobado: 24 de agosto de 2014
Contacto para la correspondencia: Castillo González William. E-Mail: revmedme@infomed.sld.cu
Laboratorio central de líquido cefalorraquídeo (LABCEL). Ramón Pinto No 202. 10 de Octubre. La Habana, Cuba.

