










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Salud Animal
versión impresa ISSN 0253-570X
Rev Salud Anim. v.29 n.1 La Habana ene.-abr. 2007
Artículo original
CLONAJE MOLECULAR Y EXPRESIÓN DEL GEN QUE CODIFICA PARA LA PROTEÍNA ESTRUCTURAL DEL VIRUS DE LA ENFERMEDAD INFECCIOSA DE LA BOLSA, VP3
MOLECULAR CLONING AND EXPRESSION OF THE GENE THAT CODIFIES FOR THE INFECTIOUS BURSAL DISEASE VIRUS STRUCTURAL PROTEIN, VP3
J.A. Agüero*, Yanaysi Ceballo**, Yadmell González*, Siomara Martínez* y G.A. Enríquez**
*Centro Nacional de Sanidad Agropecuaria (CENSA), Apartado 10, San José de las Lajas, La Habana, Cuba; ** Centro de Ingeniería Genética y Biotecnología (CIGB), Ave 31 e/ 158 y 190, Playa. P.O. Box 6162. Habana 10600
RESUMEN
El virus de la enfermedad infecciosa de la bolsa (IBDV), un miembro de la familia Birnaviridae, es un virus ARN de doble cadena, capaz de causar considerables pérdidas económicas al inducir destrucción de la bolsa, inmunosupresión y alta mortalidad en pollos jóvenes. La cápsida del virus está formada por las proteínas estructurales VP2 y VP3. Se amplificó por PCR el gen vp3 (870pb), a partir del vector pcDNA3-Poly que contiene el gen que codifica para la poliproteína (pVP2-VP4-VP3) del virus. El producto de PCR se clonó en el plásmido pQE30 y se expresó en la cepa M15 de E. coli a altos niveles como una proteína de fusión con una cola de hexahistidina. Así se obtuvo una proteína antigénicamente activa, como se demostró por Western-Blot con un anticuerpo policlonal contra el virus, con un peso molecular aproximado de 32kDa. Se describió un protocolo para extraer la proteína en forma soluble, acoplado a un eficiente sistema de purificación en un solo paso, basado en la cromatografía de afinidad por metales inmovilizados, mediante el cual se obtuvo un producto con un 90% de pureza. En conejos se generaron sueros de altos títulos de anticuerpos contra la VP3 recombinante capaces de reconocer tanto a la proteína recombinante, como a la proteína nativa del virus. Son necesarios experimentos posteriores para evaluar el potencial de estos anticuerpos en el monitoreo de la expresión de la VP3 o la poliproteína del virus en otros sistemas heterólogos.
Palabras clave: VP3; IBDV; cápsida; proteína recombinante; IMAC.
ABSTRACT
Infectious bursal disease virus (IBDV), a member of the Birnaviridae family, is a double stranded RNA virus that causes considerable economic losses in the poultry industry by inducing bursal destruction, immunosuppression and high mortality in young chickens. Viral capsid is formed by two structural proteins, VP2 and VP3. The vp3 gene (870pb) was amplified by PCR from the vector pcDNA3-Poly containing the gene that encodes for virus polyprotein (pVP2-VP4-VP4). The PCR product was cloned into pQE30 vector. The cloning was screened by PCR using vp3 primers. The gene was expressed as a his-tag fusion protein in M15 E. coli strain. The recombinant protein molecular weight was about 32kDa. The protein was recognized by Western-Blot using an antibody against the virus. Using an ion metal affinity chromatography procedure, a 90% pure recombinant product was obtained. In rabbits, high-titer antibodies against the recombinant VP3, able of recognizing both, the recombinant and native VP3, were obtained. The potential use of these antibodies for VP3 or virus polyprotein expression screening in heterologous systems should be further evaluated.
Key words: VP3; IBDV; capsid; recombinant protein; IMAC.
INTRODUCCIÓN
El virus de la enfermedad infecciosa de la bolsa (IBDV) es el miembro prototipo del género Avibirnavirus en la familia Birnaviridae (15), un importante patógeno de los pollos, responsable de importantes pérdidas económicas en la industria avícola mundial y uno de los principales peligros para varias especies de pájaros salvajes (7). Se han identificado dos serotipos, I y II (11). Todas las cepas patógenas de IBDV que se conocen, pertenecen al serotipo I, mientras que los virus del serotipo II, capaces de infectar pollos y pavos, no provocan manifestaciones clínicas de la enfermedad (10). El IBDV se multiplica rápidamente en la bolsa de Fabricius, en linfocitos B en desarrollo, provocando inmunosupresión. Esto incrementa la susceptibilidad a las infecciones por patógenos oportunistas y reduce las tasas de crecimiento de los animales sobrevivientes (2). Las vacunas clásicas pueden ser incapaces de conferir protección contra cepas altamente virulentas, como se puso de manifiesto durante dos brotes ocurridos en la pasada década (25). Solo mediante la comprensión de la biología molecular del IBDV, incluyendo la estructura viral será posible avanzar en el control de la enfermedad.
El IBDV es un virus icosahédrico no cubierto de aproximadamente 60 nm de diámetro (8). Su genoma está compuesto de dos segmentos de ARN de doble cadena designados A y B (20). El segmento B codifica para VP1, una proteína de 95 kDa que comparte algunas semejanzas en la secuencia primaria con ARN-polimerasa de diversos orígenes (5). El segmento mayor A (3.3kb), contiene dos marcos de lectura abiertos (MLA), parcialmente solapados. El primer MLA codifica para la proteína no estructural VP5 (17kDa), cuyas propiedades funcionales no están aún claras (17). El segundo MLA codifica para una poliproteína de 110kDa (pVP2-VP4-VP3) (9), la cual es digerida autoproteolíticamente y da lugar a tres proteínas: pVP2 (48kDa), VP3 (32kDa) y VP4 (28kDa). VP4 es responsable de este autoprocesamiento de la poliproteína (12). VP2 y VP3 son las proteínas estructurales principales del virión. Aunque algunos autores plantean que VP2 no es el único determinante de fenotipos altamente virulentos (3), sí está demostrado que esta proteína resulta decisiva en la patogenicidad del IBDV (21; 26). Por su parte VP3 se encuentra formando parte de la capa interior del virión (4) y contiene una región carboxilo-terminal altamente básica que interacciona con el ARN empacado (9).
La naturaleza inmunosupresora del virus (13) dificulta en ocasiones la generación de anticuerpos con el título necesario, a partir de animales de campo. Otra limitante de los anticuerpos obtenidos, a partir del antígeno completo es que su especificidad no siempre es la deseada. La carencia de anticuerpos con alta especificidad y afinidad por los antígenos del IBDV puede dificultar y retrasar en algunos casos las investigaciones relacionadas con este agente. En nuestro caso particular, en el cual se requiere detectar cantidades muy bajas de los productos del gen de la poliproteína, la carencia de estos anticuerpos se ha convertido en una gran limitante.
Para superar estas limitaciones algunos autores han escogido la vía de la obtención de anticuerpos, a partir de antígenos sintéticos o recombinantes (6). El empleo de sistemas heterólogos para la expresión recombinante de las proteínas del IBDV en sistemas celulares variados se ha descrito ampliamente. Estos estudios han tenido como objetivo la comprensión de la morfogénesis y la estructura del virus. Así se estudió la expresión de la poliproteína en E.coli (1; 12). En levadura se estudió la interacción de proteínas homólogas y heterólogas del virus (23). Recientemente se ha demostrado que la expresión de la poliproteína en células de mamíferos (6) o de insecto (24), mediante el empleo de vectores virales recombinantes conduce a la formación de partículas similares a virus (PSV) con una talla y morfología idéntica a la de los viriones del IBDV. Este resultado se obtuvo, por último, en células vegetales, con la formación de PSV en plantas.
En este trabajo se describe el clonaje molecular del gen que codifica para la proteína estructural de la cápsida (VP3) del virus de la cepa de campo Soroa, en un vector de expresión en E. coli, el pQE30, así como su expresión, purificación y la obtención de anticuerpos de alta afinidad y especificidad por esta proteína.
MATERIALES Y MÉTODOS
Vectores, cepas bacterianas y condiciones de cultivo. Las células XL1 Blue competentes, así como las M15 competentes se obtuvieron del banco de cepas del Centro de Ingeniería Genética y Biotecnología (CIGB). El plásmido pcDNA3-poly contentivo del gen de la poliproteína de la cepa Soroa del IBDV se obtuvo del banco de plásmidos del grupo de Biología Molecular del CENSA. El vector pQE30 se purificó a partir de la cepa DH5a de E.coli previamente transformado con este vector. Las cepas Escherichia coli DH5a y XL1-Blue transformadas con los plásmidos pQE30 y pQE30 recombinantes respectivamente, se crecieron en medio Luria (LB) líquido y agar suplementado con ampicillina (100µg/mL), a 37°C toda la noche, con agitación a 200rpm, en el caso de los cultivos líquidos.
Preparación del vector. El plásmido pQE30 se purificó empleando el Wizard Plus SV Minipreps DNA Purification System (Promega), siguiendo el protocolo descrito por el fabricante. Se digirieron 5mg del vector con 50u de KpnI (Promega), en un volumen total de 100mL y en presencia de BSA 0.1mg/mL y 10mL de tampón J 10x (Promega), la digestión se realizó durante 3 horas a 37°C. El producto de digestión se aplicó en un gel de agarosa de bajo punto de fusión al 0.8%, en buffer TB, se separó la banda del vector digerido y se purificó usando el QIAquick Gel Extraction Kit (QIAGEN). La segunda digestión se realizó con 50u de SalI (Promega) en un volumen total de 100mL y en presencia de BSA (Promega) 0.1mg/mLy 10 mL de tampón D 10x (Promega), la misma se ejecutó por 3h a 37°C. La enzima SalI se inactivó incubando la mezcla de reacción 15 min a 65°C. El producto final se purificó mediante el QIAquick Gel Extraction Kit (QIAGEN) según protocolo del fabricante.
Generación del plásmido recombinante. El gen vp3 fue obtenido por PCR usando el plásmido pcDNA3-Poly como molde y los cebadores VP3S TCGGTACCCGTTTCCCTCACAATCCACGCGACTG (sentido) y VP3AS CCGTCGACCACTCAAGGTCCTCATCAGAGACGG (antisentido). El gen se amplificó adicionando 500ng del plásmido a la mezcla de PCR que contenía 5mL de tampón de la Pfu ADN polimerasa 10x (Promega), dNTPs (Promega) a 200mM, ambos cebadores a 250nM y 0.5mL (2-3 u/mL) de Pfu (Promega) en un volumen total de 50mL. El programa de PCR se corrió a las siguientes condiciones: 1 ciclo de 95°C por 2 min, seguido por 35 ciclos de 95°C por 1 min, 60°C por 1 min y 72°C por 2 min y por último un ciclo de 72°C por 5 min.
El producto de PCR se purificó empleando el Kit de QIAGEN (QIAquick Gel Extraction), 2.5mg del producto de PCR se digirieron con 25u de KpnI (Promega) en un volumen total de 100mL, en presencia de BSA 0.1mg/mL (Promega) y 10mL de tampón J 10x (Promega), a 37°C durante 3h. El producto de digestión se purificó usando el QIAquick Gel Extraction Kit de QIAGEN. La segunda digestión se realizó con 25u de SalI (Promega) en un volumen de 100mL, en presencia de BSA 0.1mg/mL (Promega) y 10mL de tampón D 10x (Promega). Para obtener la construcción pQE30-vp3 se ligó el producto de PCR con el vector pQE30 previamente digerido. La concentración del vector en la mezcla de digestión fue de 14.6 ng/mL y la del inserto de 14.4 ng/mL. Se emplearon 4u de T4 ligasa (Promega), 2mL de tampón T4 ligasa 10x (Promega) en un volumen total de 20mL y la ligazón se realizó a 22°C, durante 5h.
Para la transformación se emplearon células Ca2+ competentes de la cepa XL1-Blue de E. coli y se siguió el siguiente protocolo: Se adicionaron 10mL del producto de ligazón a 150mL de células competentes, se incubó en hielo 20 min,, luego a 42°C 2 min y por último de nuevo en hielo 5´. Se añadió 1mL de medio LB y se incubó 1h a 37°C a 50rpm. Por último, el cultivo se sembró en placas LB con ampicillina a 100mg/mL y se incubó toda la noche a 37°C.
Las colonias que contenían los plásmidos recombinantes se identificaron por PCR. Para esto se sembraron las colonias en LB suplementado con ampicillina (100µg/mL) y se incubaron toda la noche a 37°C. Se extrajeron 100mL de los cultivos, se centrifugaron 5 min a 10000 rpm y se reconstituyeron en 100mL de H2O, la suspensión se calentó 10 min a 95°C y se volvió a centrifugar 5 min a 10000 rpm. Siguiendo el protocolo descrito previamente se le realizó PCR a 5mL de cada uno de los sobrenadantes.
Expresión de la VP3 recombinante. Se transformaron células competentes de la cepa de expresión M15 de E.coli con la construcción pQE30-vp3 obtenida. La cepa recombinante se cultivó toda la noche a 37°C en medio LB suplementado con ampicillina (100µg/mL) y kanamicina (25µg/mL). Al día siguiente, a 1L de LB suplementado con ampicillina (100µg/mL) y kanamicina (25µg/mL), se le adicionaron 20mL del inóculo de toda la noche. El cultivo se incubó por aproximadamente 2h 30min a 37°C hasta que alcanzó una DO a 600nm entre 0.7 y 0.9. En este momento, la expresión de VP3 se indujo con la adición de IPTG hasta una concentración final de 2mM. Después de 5 horas de incubación a 37°C, las células se recolectaron por centrifugación 10 minutos a 6000rpm y se conservaron a _20°C.
Purificación de VP3 por cromatografía de afinidad por iones metálicos. El precipitado celular obtenido de 1L de cultivo se resuspendió en 25mL de una solución de Tris 50mM pH 7.5 y las células se rompieron por lisis alcalina, adicionando 25mL de una solución de NaOH 200mM, SDS 1%, e incubando a Tamb por 20 minutos. Al lisado se le adicionaron 50mL de tampón Tris 10mM_NaH2PO4 100mM pH 5 y se centrifugó 30 minutos a 12000rpm. El sobrenadante se filtró por un prefiltro de fibra de vidrio y un filtro de 0.45µm. Se cargó una columna de Chelating Sepharose Fast Flow (Pharmacia Biotech) de 4mL, con 8mL de NiSO4 100mM. La columna se lavó con 5 volúmenes de H2O y posteriormente se equilibró con 5 volúmenes de tampón de inicio (Tris 10mM _ NaH2PO4 100mM pH 8). En la columna se aplicaron 25mL de la muestra clarificada ajustada a pH 8. Se lavó la columna con 3 volúmenes de tampón de inicio, seguidos de 3 volúmenes de tampón de lavado (Tris 10mM _ NaH2PO4 100mM pH 6.5). La proteína VP3 se eluyó con tampón de elusión (Tris 10mM _ NaH2PO4 100mM pH 5.9). Todas las soluciones se pasaron por filtros de 0.45µm. El proceso se desarrolló con un flujo de 1mL/min.
Se extrajeron alícuotas de 10µL de las fracciones de 4mL y se analizaron por SDS-PAGE. El porciento de la proteína recombinante en cada fracción se determinó por análisis densitométrico de los geles teñidos con Coomasie Brilliant Blue R-250 empleando el programa de análisis de geles Molecular Analyst.
Obtención de anticuerpos contra VP3 recombinante. Se inmunizaron dos conejos gigantes blancos según el siguiente esquema: Inmunización con 200µg de VP3 purificada en 500µL empleando adyuvante completo de Freund, por vía subcutánea; a los 14 días se administraron 100µg de proteína en 500µL con adyuvante incompleto de Freund (AIF) (250µL por vía subcutánea y 250µL por vía intradérmica); en el día 28 100µg en 500µL por vía subcutánea en AIF; en el día 56, 50µg en 500µL subcutáneamente en AIF. Los animales fueron desangrados al día 70, después de la primera inoculación. La titulación de los sueros se realizó por ELISA definiendo el título como la mayor dilución la DO del control positivo (VP3) se encontraba por encima de 1 mientras la DO del blanco no sobrepasaba el valor de 0.1. También se caracterizó por Western-Blot el reconocimiento de la VP3 (recombinante y viral) por parte del suero.
Análisis por SDS-PAGE y Western-Blot. La electroforesis se realizó bajo condiciones reductoras y se corrió a corriente constante con 60mA en un gel de acrilamida al 10% y al 12%. Para el Western-Blot se hizo una transferencia semiseca de las proteínas. La membrana se bloqueó por una hora en una solución de PBS-BSA 1% y luego se incubó 2 horas a 37°C con el anticuerpo policlonal contra IBDV (SIAFA) diluido 1/500 en PBS-BSA 1%. Seguidamente se incubó 1 hora a 37°C con el conjugado anti IgG de Pollo _ Fosfatasa Alcalina (Sigma) y por último la reacción se reveló con NBT-BCIP (Promega).
RESULTADOS Y DISCUSIÓN
Se escogió la vía de obtener los anticuerpos, a partir de antígenos recombinantes porque una vez que se generaran los clones recombinantes contaríamos con una fuente estable de los mismos para, no solo obtener los anticuerpos, sino también para ser empleados con otros fines como puede ser el desarrollo de sistemas de diagnóstico y el contar con antígenos controles en los sistemas de monitoreo de la expresión de la poliproteína.
La región que codifica para la proteína VP3 se amplificó por PCR a partir de la construcción pcDNA3-Poly, donde previamente había sido clonada la poliproteína del virus. A los cebadores sentido y antisentido se les incluyeron los sitios de restricción KpnI y SalI respectivamente para facilitar los pasos posteriores de clonaje. Los productos de PCR, cuya talla se correspondió con la talla correcta de aproximadamente 870pb (resultados no se muestran) de la VP3 viral, se purificaron y se clonaron en el vector pQE30 para generar la construcción pQE30-vp3.
La transformación inicial, que se efectuó con propósitos de monitoreo, se realizó en células competentes XL1 Blue, debido a que esta cepa facilita la propagación y posterior purificación de los plásmidos recombinantes. Sin embargo la proteína clonada en el plásmido pQE30-vp3 debe ser expresada en la cepa M15 de E. coli (o en la cepa SG13009) que, además de permitir altos niveles de expresión, garantiza un alto grado de control de la misma. En estas cepas la expresión regulada por el promotor T5 del fago de E. coli y las dos secuencias del operador lac que posee el plásmido pQE30 es extremadamente eficiente y solo puede ser evitada por la presencia de altos niveles del represor lac. La cepa M15 contiene múltiples copias del plásmido pREP4, el cual posee el gen lacI que codifica para el represor lac. Cepas de E.coli que contengan el gen lacIq, como JM109, TG1 y XL1-Blue, también pueden ser empleadas como hospederos de expresión, ya que en este gen el promotor ha sido mutado y produce 10 veces más represor lac que el gen salvaje, aunque estas van a ofrecer una expresión no tan sólidamente regulada como las cepas poseedoras del plásmido pREP4. Por esta razón se aisló el plásmido del clon 19 (uno de los 6, de 98 clones tomados al azar de pQE30-vp3 en XL1-Blue, que resultaron positivos cuando se monitorearon por PCR con los cebadores VP3S y VP3AS, específicos para el gen vp3, al amplificarse un fragmento de 870pb) y con este se transformaron células competentes de la cepa de expresión M15.
Como resultado de la inducción con IPTG de un cultivo de esta última cepa transformada se detectó por SDS-PAGE la aparición de una banda de aproximadamente 32kDa que fue reconocida por el anticuerpo policlonal específico contra IBDV por Western Blot (Fig.1).
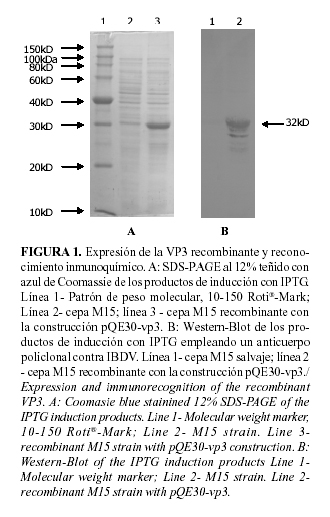
En este resultado un hecho resultó interesante y significativo; otra proteína, con una talla entre 2.5 y 3kDa menos que la de 32kDa, también era reconocida por el suero contra IBDV. Teniendo en cuenta que esta reacción no se observa con las proteínas de la cepa de expresión M15 (Fig. 1), es de suponer que la proteína de aproximadamente 29kDa sea también un producto codificado por la construcción recombinante obtenida por nosotros, pQE30-vp3. Tales formas de VP3 con una talla menor a la esperada, que pueden incluso llegar a ser dos (23), son frecuentemente observadas en células infectadas con el virus, pero usualmente se ignoran o se confunden con VP4. Estos dobletes de VP3 también han sido observados en sistemas recombinantes donde se ha expresado VP3 (18; b), e incluso donde esta proteína, ha sido expresada fusionada a seis histidinas, en células de insecto usando baculovirus como vector (14; 19). Aunque algunos autores sugieren que esto podría estar generado por una iniciación interna de la translación en un codón AUG localizado 15 residuos después del codón de iniciación (18), los estudios realizados apuntan a que el hecho se debe a una digestión proteolítica en el extremo C-terminal de la proteína, que puede estar generada por proteasas, tales como caspasas, o por una actividad autoproteolítica de la VP3 (14; 19). A pesar de que la naturaleza y la función de estas formas permanecen sin conocerse, es interesante que solo la VP3 de talla completa se incorpora al virión, sugiriendo que las formas más cortas pueden tener funciones no estructurales adicionales (14). Si realmente la causa de este fenómeno está dada por la actividad de alguna proteasa entonces la proteólisis pudiera minimizarse mediante la adición de algún inhibidor de estas como el PMSF, o de EDTA que es capaz de acomplejar iones metálicos importantes en la actividad de algunas enzimas. En nuestro caso la segunda no es una solución aconsejable porque la presencia de EDTA interfiere en los sistemas de purificación por IMAC como el que será empleado por nosotros posteriormente para la purificación de la proteína.
Estos resultados demostraron que se había obtenido un clon recombinante con la construcción pQE30-vp3, capaz de expresar la VP3 de una manera antigénicamente funcional.
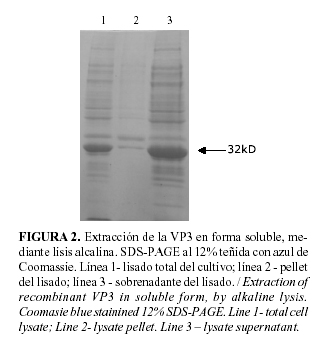
Cuando las proteínas recombinantes se expresan a altos niveles, usualmente precipitan en una forma insoluble (cuerpos de inclusión) (22; 16). Para cualquier uso terapéutico posterior estas deben ser renaturalizadas in vitro. Aunque se han descrito varios protocolos para facilitar la producción de proteínas solubles en sistemas procariotes, la mayoría de ellos son laboriosos y requieren de una significativa experiencia técnica (22; 16). El protocolo de ruptura celular empleado en este trabajo permitió extraer la proteína recombinante de manera soluble (Fig.2), sin necesidad de adicionar urea al medio, evitando los inconvenientes que esto puede traer por lo agresivo de este reactivo y las dificultades que puede implicar el proceso de renaturalización de una proteína disuelta en presencia de urea. La electroforesis muestra que una pequeña parte de la proteína recombinante aún permanece en la fracción insoluble, pero esta pérdida se compensa por las ya mencionadas ventajas de la solubilización en ausencia de urea. Es posible, no obstante, que incrementando el tiempo de lisis o aumentando el volumen en que la misma se realiza puedan disminuirse la pérdidas por este concepto.
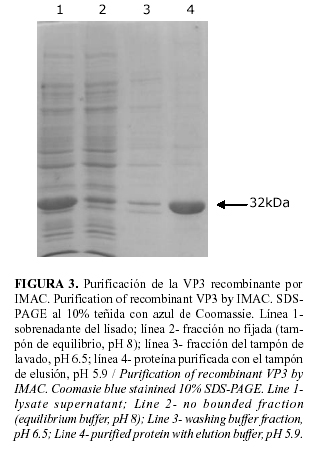
El plásmido seleccionado para el clonaje, el pQE30, es un plásmido de fusión que contiene la secuencia que codifica para seis histidinas ubicadas en el extremo 3´ del sitio de clonaje, que facilitan la posterior purificación de la proteína de fusión resultante, mediante cromatografía de afinidad por iones metálicos (IMAC). La proteína inmovilizada puede ser eluida de la columna de dos maneras diferentes, mediante una elusión no competitiva (decrecimiento de pH) o por elusión competitiva (incremento de la concentración de imidazol en el tampón de lavado). En nuestro caso la selección de la primera variante evitó pasos posteriores para la eliminación del imidazol, así como disminuyó los costos de nuestro proceso, sin afectar la efectividad del mismo. En nuestras condiciones, empleando el gradiente de pH, fue posible eluir la proteína con un alto nivel de pureza, aproximadamente de un 90% (Fig. 3) y con un pH (5.9) alejado de los valores más ácidos que hubieran sido necesario emplear de haber realizado el proceso en presencia de urea. En estudios previos, la lisis celular se había realizado en presencia de urea 8M y la proteína era eluída de la columna, con pH 4 (resultados no se muestran). Esto puede deberse a que en presencia de altas concentraciones de un agente caotrópico la proteína se encuentra totalmente desnaturalizada y sus histidinas mucho más accesibles a los iones metálicos de la columna, por lo que su fijación a la matriz es mucho más fuerte y son necesarias condiciones más drásticas para poder eluir la proteína fijada.
El esquema seleccionado para inmunizar los animales resultó muy efectivo, ya que la respuesta fue elevada, en lo cual influyó la calidad del antígeno con el que se preparó el inmunógeno. Como se puede observar en la Figura 4, el título de los sueros por ELISA, alcanzó el valor de 1:500 000, al ser esta la mayor dilución en la cual la DO de la reacción con VP3 es mayor que 1 y la del blanco no supera el valor de 0.1. Este título es muy superior a los títulos de los sueros contra IBDV con los que contábamos hasta ese momento en el laboratorio, los cuales no superaban 1:1000. Este resultado nos hace pensar que este suero será muy eficiente para detectar VP3, incluso cuando la proteína se encuentre en muy bajas concentraciones en la muestra.

Otro aspecto importante a tener en cuenta para evaluar la calidad del suero obtenido, es el grado de especificidad que presenta, entendido en este caso como su nivel de reconocimiento de las proteínas de E. coli, por ser esta la única fuente de proteínas contaminantes que tuvo el antígeno que se utilizó en la inmunización de los animales. La técnica empleada para este fin, el Western-Blot (resultados no se muestran), permitió observar que el nivel de reacción con las proteínas de la bacteria es prácticamente nulo, a una dilución del mismo que permite detectar inequívocamente a VP3. Las proteínas adicionales que se detectan en la muestra de la cepa recombinante pueden deberse a cierto nivel de degradación de la VP3, aspecto este que fue previamente discutido. Queda por evaluar entonces el grado de reacción cruzada de este suero con proteínas de otras fuentes. En nuestro caso y debido a nuestras investigaciones actuales, sería muy importante realizar este estudio con extractos de plantas de tabaco, por ser este el sistema donde experimentamos la expresión de la poliproteína del IBDV.
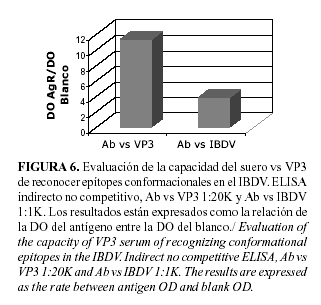
Por último, y para confirmar la calidad del suero obtenido, desarrollamos un Western-Blot que nos permitió evaluar si el mismo era capaz de reconocer a la VP3 viral. Los resultados de este experimento (Fig. 5) revelan que los anticuerpos obtenidos reaccionan con una banda de 30kDa y otra de 28kDa (previamente ya se discutió acerca de la presencia de estos dobletes) del antígeno viral que se corresponden con la talla de la VP3. Esta evaluación la realizamos entonces por ELISA para conocer si nuestros anticuerpos eran capaces solo de reconocer epítopes lineales o si poseían la capacidad de hacerlo también con los conformacionales. La Figura 6 muestra como el reconocimiento del antígeno por nuestro suero es superior al que se logra con el suero de campo (la señal es tres veces mayor). Este resultado, sobre todo si tomamos en cuenta que el suero de campo estaba diluido 1:1000, mientras que el nuestro lo estaba 1:20 000, demuestra que estos últimos son capaces de reconocer dominios conformacionales y que además, tienen una alta afinidad por la VP3 del antígeno viral. Esto demostró que los sueros obtenidos, a partir de la VP3 recombinante presentaron una alta afinidad y especificidad por esta proteína, lo que los convierte en candidatos ideales para desarrollar sistemas analíticos de alta especificidad y sensibilidad, capaces de detectar con alto grado de confiabilidad la expresión de la misma.

CONCLUSIONES
Como conclusiones podemos decir que el protocolo de lisis alcalina empleado para la ruptura celular y la extracción de proteínas, combinado con la purificación por IMAC con gradiente de pH, permite obtener la proteína recombinante VP3 de la cápsida viral del virus de la enfermedad infecciosa de la bolsa con un alto nivel de pureza, y conservando su actividad antigénica e inmunógena. Además, el suero obtenido contra la VP3, debido a su especificidad, su alto título, y su capacidad de reconocer tanto a la VP3 recombinante como a la viral, es un excelente candidato para desarrollar un sistema analítico altamente específico y sensible para detectar la presencia de esta proteína.
Tanto la proteína VP3 recombinante, como los anticuerpos contra la misma obtenidos en nuestro trabajo, pueden tener importantes aplicaciones en las investigaciones relacionadas con la obtención de una vacuna contra el IBDV. La proteína, más allá de su empleo en la obtención de anticuerpos que hemos discutido, tiene un gran valor per se,ya que puede ser usada como control positivo en los ensayos inmunoquímicos destinados a la detección de las proteínas del virus. También podría tener una gran aplicación en el desarrollo de sistemas de diagnóstico complementarios a vacunas basadas en VP2, que permitirían diferenciar entre animales vacunados y los naturalmente infectados por el virus.
REFERENCIAS
1. Azad AA, Jagadish MN, Brown MA, Hudson PJ. Deletion mapping and expressión in Escherichia coli of the large genomic segment of birnavirus. Virol. 1987;161:145-152.
2. Becht Hand Muller H. Infectious bursal disease - B cell dependent immunodeficiency syndrome in chickens. Behring Institute Mitteilungen. 1991;217-225.
3. Boot HJ, Ter Huarne AAHM, Hoekman AJW, Peeters BPH, Gielkens ALJ. Rescue of very virulent and mosaic infectious bursal disease virus from cloned cDNA: VP2 is not the sole determinant of the very virulent phenotype. J Virol. 2000;74(15):6701-6711.
4. Bottcher B, Kiselev NA, Stel´Mashcheck VJ, Perevozchikova NA, Borisov AV, Crowther RA. Three-dimensional structure of infectious bursal disease virus determined by electron cryomicroscopy. J Virol. 1997;71:325-330.
5. Bruenn JA. Relationships among positive strand and double-strand RNA viruses as viewed through their RNA-dependent RNA polymerases. Nucleic Acids Res.1991;19:217-226.
6. Fernández A, Risco C, Martínez S, Albar JP, Rodríguez JF. Expression of infectious bursal disease virus results in the formation of virus-like particles. J Gen. Virol. 1998;79:1047-1054.
7. Gardner H, Kerry K, Riddel M, Brouweer S, Gluson L. Poultry virus infection in Antarctic penguins. Nature. 1997;387:245.
8. Hirai K, Shimakura S. Structure of infectious bursal disease virus. J. Virol. 1974;14:957-964.
9. Hudson PJ, Mc Kern NM, Power BE, Azad AA. Genomic structure of the large RNA segment of infectious bursal disease virus. Nucleic Acid Research. 1986;14:5001-5012.
10.Ismail NM, Saif YM, Moorhead PD. Lack of pathogenicity of five serotype 2 infectious bursal disease viruses in chickens. Avian Dis. 1988;32:757-759.
11.Jackwood DJ, Saif YM. Antigenic diversity of infectious bursal disease viruses. Avian Dis.1987;31:766-770.
12.Jagadish MN, Staton VJ, Hudson PJ, Azad AA. Birnavirus precursor polyprotein is processed in Escherichi coli by its own virus- encoded polypeptide. J Virol. 1988;62:1084-1087.
13.Kim IJ, You SK, Kim H, Yeh HY, Sharma JM. Characteristics of bursal T lymphocytes induced by infectious bursal disease virus. J Virol. 2000;74(19):8884-8892.
14.Kochan G, González D, Rodríguez JF. Characterization of the RNA-binding activity of VP3, a major structural protein of infectious bursal disease virus. Arch Virol. 2003;148:723-744.
15.Leong JC, Brown D, Dobos P, Ludert JE, Muller H, et al. Family Birnaviridae. 2000;p 481-490. In MHV van Regenmortel, CM Fanguet, DHL Bishop, EB Carstens, MK Estes, SM Lemon, J Manilogg, MA Mayo, DJ McGeoch, CR Pringle, RB Wickner (ed.), Virus taxomy: classification and nomenclature of viruses. Seventh Report of the International Committee on Taxonomy of Viruses. Academic Press, San Diego, California.
16.Li M, Su ZG, Jarson JC. In vitro protein refolding by chromatographic procedures. Protein Expr Purif. 2004;33(1):1-10.
17.Lombardo E, Maraver A, Espinosa I, Fernández A, Rodríguez JF. VP5, the nonstructural polypeptide of infectious bursal disease virus, accumulates within the host plasma membrane and induces cell lysis. Virol. 2000;277:345-357.
18.Maraver A, Clemente R, Rodríguez JF, Lombardo E. Identification and molecular characterization of the RNA polymerase-binding motif of infectious bursal disease virus inner capsid protein VP3. J Virol. 2003;77(4):2459-2468.
19.Maraver A, Oña A, Abaitua F, González D, Clemente R, Ruiz-Díaz JA, et al . The oligomerization domain of VP3, the scaffolding protein of infectious bursal disease virus, plays a critical role in capsid assembly. J Virol. 2003;77(11):6438-6449.
20.Muller H, Scholtissek C, Becht H. The genome of infectious bursal disease virus consists of two segments of double- stranded RNA. J Virol 1979;31: 584-589.
21.Mund E. Tissue culture infectivity of different strains of infectious bursal disease virus is determined by distinct amino acids in VP2. J Gen Virol. 1999;80:2067-2076.
22.Platis D, Foster GR. High yield expression, refolding, and characterization of recombinant interferon alpha2/alpha8 hybrids in Escherichia coli. Protein Expr Purif. 2003;31(2):222-30.
23.Tacken MGJ, Rottier PJM, Gielkens ALJ, Peeters BPH. Interactions in vivo between the proteins of infectious bursal disease virus: capsid protein VP3 interacts with the RNA-dependent RNA polymerase, VP1. J Gen Virol. 2000;81:209-218.
24.Vakharia VN. Development of recombinant vaccines against infectious bursal disease. Biotech Annu Rev. 1997;3:151-168.
25.Van der Berg TP. Acute infectious bursal disease in poultry: a review. Avian Pathol. 2000;29:175-194.
26.Van Loon AAWM, de Hass N, Zeyda I, Mundt E. Alteration of amino acids in VP2 of very virulent infectious bursal disease virus results in tissue culture adaptation and attenuation in chickens. J Gen Virol. 2002;83:121- 129.
(Recibido 8-11-2005; Aceptado 13-2-2006)


{kind=link}