










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.77 n.1 Ciudad de la Habana ene.-mar. 2005
Centro Nacional de Genética Médica
Fenotipo turneriano asociado al cromosoma Y en anillo
Dra. Estela Morales Peralta,1 Lic. Carlos Viñas Portilla,2 Jeorges Pérez Giardinú3 y Lic. Teresa Collazo Mesa4
Resumen
El síndrome de Turner es una enfermedad que típicamente afecta a las hembras. En nuestro trabajo describimos un paciente con los signos principales de esta. Su cariotipo fue 46, X r(Y) /45, X. Este mosaicismo se explica por la inestabilidad del anillo cromosómico que conduce a su pérdida luego de la mitosis. Mediante pruebas moleculares, que incluyeron la identificación de los genes SRY y AM-XY, obtuvimos los resultados habituales encontrados en varones. De estos hallazgos podemos concluir que el material genético perdido, como parte del proceso de formación del anillo cromosómico, es distal a Y p11.3. Esto demuestra que los genes anti-Turner se encuentran localizados en esta región pseudoautosómica.
Palabras clave : Síndrome de Turner, cromosoma Y, cromosoma en anillo.
El síndrome de Turner es una enfermedad genética que afecta generalmente a las hembras. Presenta una frecuencia de 4 cada 10 000 recién nacidas vivas.1 Su cuadro clínico se caracteriza por una variedad de anomalías: esqueléticas, linfoedémicas y gonadales , que conforman el conocido fenotipo turneriano. Se asocia con la ausencia de todo, o parte, del segundo cromosoma X.2 En este trabajo describimos a un paciente varón con un síndrome por deleción, producido por un cromosoma Y en anillo, que presenta los signos principales descritos en el síndrome de Turner.
PRESENTACIÓN DEL CASO
Se trata de un paciente masculino, fruto del primer embarazo de 35,3 semanas de gestación, que evolucionó con sepsis urinaria e hipertensión arterial. En el momento del nacimiento sus padres tenían edad adecuada para la reproducción (23 años él y 20 años ella). El parto fue distócico, con uso de forceps; el peso al nacimiento fue de 2 730 g, circunferencia cefálica de 34 cm y talla de 49 cm. Se refiere que presentó retardo del crecimiento, siendo siempre de una talla menor que la de sus contemporáneos. Su desarrollo psicomotor y desempeño intelectual fueron normales. Al examen físico realizado a los 10 años presentó talla de 117 cm (menor del tercer percentil) y peso de 24 kg (relación peso /talla: 97 percentil),3 normocráneo, cara triangular con desviación antimongoloidea de las hendiduras palpebrales, boca en sombrero de tres picos; orejas grandes, prominentes y con rotación posterior; cuello corto con pterigium colli, limitación de los movimientos e implantación baja del pelo en la nuca; tórax en escudo con mamilas separadas, numerosos nevos pigmentarios, cubitus valgus, braquidactilia, clinodactilia de los quintos dedos, hipoplasia de las uñas; separación entre el primero y segundo dedos del pie; hipospadia penoescrotal y criptorquídea derecha.
Mediante ultrasonido renal se encontró mala rotación de los riñones. En el estudio radiológico de la columna vertebral se observó fusión de vértebras C2 y C3 y bloque constituido por C4, C5 y C6, con marcado estrechamiento de los espacios intervertebrales, hemivértebra a nivel de T2 y apéndice costiforme en L1 .
A los 20 años se revalúa el caso y se encuentran en su evolución los siguientes datos: talla de 145 cm (por debajo del tercer percentil para la edad ), peso de 30 kg y características sexuales masculinas normales (figura 1).
Fig. 1. Cuadro clínico con características sexuales secundarias masculinas normales

A: Cuello corto con implantación del cabello baja en la nuca y orejas despegadas.
B: Mamilas separadas.
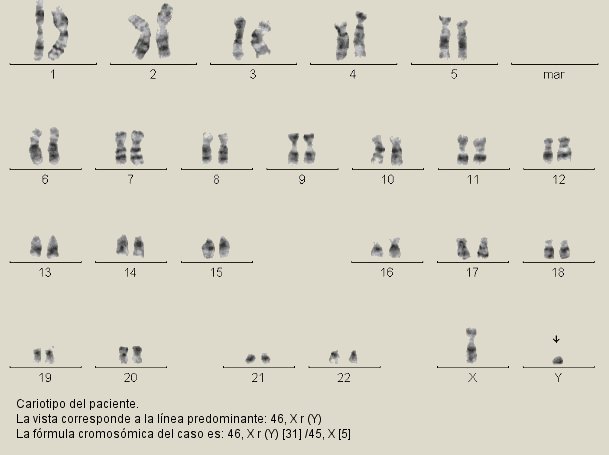
Los cromosomas, estudiados por microtécnica de cultivo sin suplemento exógeno, se identificaron mediante bandas G. Se concluyó que el paciente presentaba un cariotipo 46, X r (Y) [31] /45, X [5] (figura 2).4, 5
Fig. 2. Cariotipo del paciente
Aplicamos para el estudio de los genes SRY y AMXY las técnicas descritas por Berta y Mehra, respectivamente.6,7 Mediante el primer procedimiento se observaron bandas de 609 y 510 pares de base (pb). Las halladas en el estudio del gen AMXY fueron de 1 000 pb, 800 pb y 470 pb, tal como se muestra en las figuras número 3 y 4.
Fig. 3. Reacción en cadena de la polimerasa para estudio del gen SRY
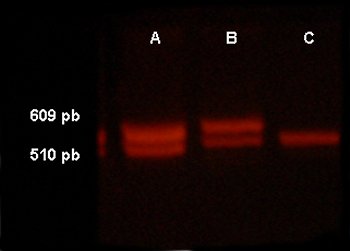
A: control varón normal; B: caso en estudio; C: control hembra normal
Las bandas observadas corresponden a 609 pb, alelo del gen SRY, y 510 pb, control interno de amplificación. (Nuestro paciente presentó las bandas correspondientes a un varón.)
Fig. 4. Reacción en cadena de la polimerasa para estudio del gen AM-XY
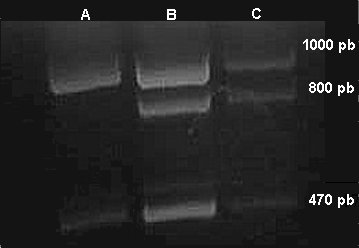
A: control hembra normal; B: caso en estudio; C: control varón normal
Las bandas observadas corresponden a 1 000 pb, alelo en el cromosoma X; 800 pb, alelo en el cromosoma Y; 470 pb, control interno de amplificación. (Es indudable que en nuestro paciente las bandas son las esperadas en un varón.)
DISCUSIÓN
Nuestro caso presenta los criterios principales para la identificación del fenotipo turneriano (tabla), el cual se caracteriza por gran variabilidad en el cuadro clínico 8 . La mayoría de los pacientes muestran algunos pero no todos los signos que se describen. El más constante es la baja talla; el resto de las manifestaciones observadas complementan el diagnóstico.
Tabla. Principales signos clínicos que habitualmente se describen en el síndrome de Turner
y los presentes en nuestro paciente
| Hallazgos clínicos en el síndrome de Turner | Frecuencia descrita | Paciente estudiado |
| Musculoesqueléticos | ||
| Talla corta | 100 | + |
| Cuello corto | 40 | + |
| Cubitus valgus | 47 | + |
| Metacarpianos / metatarsianos cortos | 37 | - |
| Escoliosis | 35 | - |
| Genu valgo | 35 | - |
| Mamilas separadas | 80 | + |
| Obstrucción linfática | ||
| Pterigium colli | 25 | + |
| Implantación baja de cabello | 42 | + |
| Edema de manos y pies | 80 | - |
| Displasia de las uñas | 13 | + |
| Defectos de células germinales | ||
| Infertilidad | 99 | ¿? |
| Otras anomalías | ||
| Cardiovasculares | 55 | - |
| Renales | 39 | + |
| Nevos pigmentados | 50 | + |
| Ptosis | 11 | - |
| Estrabismo | 18 | - |
Fuentes: http://www.aeped.es e historia clínica del paciente
Mediante del estudio cromosómico realizado comprobamos la presencia de un mosaico. En la línea celular predominante se encontró un cromosoma Y en anillo. Esta aberración cromosómica se debe a la pérdida de las regiones teloméricas de los brazos, largo y corto, del cromosoma Y, y la reunión de sus extremos.
Un cromosoma en anillo puede o no conservarse luego de la mitosis. Esto está en dependencia de que se produzca la recombinación entre las cromátides hermanas. De no efectuarse el intercambio, el cromosoma en anillo se mantiene en las células hijas. Por el contrario, si existiera recombinación se puede formar un anillo dicéntrico o dos interconectados o ensartados; estas estructuras son inestables y se pueden perder durante la migración anafásica.9 Esto puede explicar el hallazgo de la línea cromosómica 45, X observada en menor proporción, a partir del material cultivado procedente de este caso, y que es producto de un evento poscigótico.10
El método molecular más confiable para la identificación del cromosoma Y es la combinación del estudio de los genes SRY y AM-XY.11 Los resultados de la aplicación de ambas técnicas, en nuestro caso, fueron los correspondientes a un varón normal. El gen SRY, localizado en la región Yp11.3, codifica para un factor de transcripción; imprescindible en la formación de los testículos, por lo cual se le denomina factor determinante testicular; mientras el AM-XY lo hace para la síntesis de la amilogenina (AM) y está localizado en la región pericentromérica del cromosoma Y, posiblemente en Yp11. La presencia de ambos genes en el paciente estudiado, demuestra que el material perdido, como parte de la formación del anillo no incluye ninguno ellos si no una región situada hacia la zona más telomérica del brazo corto del cromosoma Y, y verifica que el cromosoma en anillo es el Y.
En los pacientes con síndrome de Turner se ha observado que aproximadamente la mitad tienen monosomía del cromosoma X (45, X), y del 5 al 10 % un isocromosoma del brazo largo de un cromosoma X (46, X, i(Xq)) que conduce a una deleción su brazo corto; en el resto predominan los mosaicos con una línea 45,X.12 Se ha determinado que la pérdida insterticial o terminal del brazo largo del cromosoma X (Xq) da como resultado baja talla y fallo ovárico primario o secundario,13 mientras la deleción del brazo corto (Xp) produce el fenotipo turneriano;14 caracterizado por la baja talla y los típicos cambios esqueléticos, como resultado de la haploinsuficiencia o simple dosis del gen SHOX (cuya denominación se deriva del inglés short stature HOmeoboX-containing gene), localizado en la región pseudoautosómica del brazo corto del cromosoma X (X pter-p22) (PAR1, del inglés pseudo-autosomic region), homóloga al locus Ypter-p11.2.15 La baja talla y el resto de las manifestaciones esqueléticas que observamos en el paciente descrito, así como los resultados de los estudios moleculares realizados sugieren la pérdida de este gen, como parte del extremo del brazo corto del cromosoma Y, que de este modo queda excluido por el proceso de formación del anillo.16
Se ha propuesto que las manifestaciones clínicas del síndrome de Turner son debido a la ausencia de los dos cromosomas X o a la haploinsuficiencia de genes de las regiones pseudoautosómicas que comparten los cromosomas X e Y.17,18 Precisamente en la zona delecionada, como resultado del cromosoma Y en anillo que presenta este paciente, están localizados genes homólogos con el cromosoma X, que pertenece al grupo que escapa a la inactivación. El cuadro clínico del paciente que describimos apoya la hipótesis de que los genes de esa región pseudoautosómica pudieran evaluarse entre los posibles, que en doble dosis, evitan el síndrome de Turner.
TURNER'S PHENOTYPE ASSOCIATED WITH RING Y CHROMOSOME
Turner's syndrome is a disease typically affecting females. In our paper, we describe a patient with its main signs. His karyotype was 46, Xr(Y)/45,X. This mosaicism is explained by the instability of the chromosomic ring leading to its loss after mitosis. By molecular tests, including the identification of SRY and AM-XY genes, we obtained the usual results found in males. According to these findings, we can conclude that the genetical material lost as part of the process of formation of the chromosomic ring is distal to Y p 11.3. This shows that the anti-Turner genes are located in this pseudoautosomal region.
Key words: Turner's syndrome, Y chromosome, ring chromosome.
Referencias bibliográficas
1. Zinn AR, Page DC, Fisher EM. Turner syndrome: the case of the missing sex chromosome. Trends Genet. 1993;9(3):90-3.
2. Tzancheva M, Kaneva R, Kumanov P, Williams G, Tyler-Smith C. Two male patients with ring Y: definition of an interval in Yq contributing to Turner syndrome. J Med Genet 1999;36(7):549-53.
3. Jordan J, Bebelagua A, Ruben M, Hernández J. Investigación nacional de crecimiento y desarrollo. Rev Cubana Pediatr 1977;49(4) : 367-390 .
4. Lantigua Cruz A, Viñas Portilla C, Delgado Oceguera E, Monte Sotolongo E del , González García N. Análisis de las aberraciones cromosómicas y del motivo de indicación en este estudio en 734 pacientes. Rev Cubana Pediatr 1989; 61(1): 44-56.
5. Seabright M. A rapid banding technique for human chromosomes. Lancet 1971; 2(7731):971-2 .
6. Berta P, Hawkins JR, Sinclair AH, Taylor A, Griffiths BL, Goodfellow PN, et al. Genetic evidence equating SRY and the testis-determining factor. Nature 1990;348(6300):448-50.
7. Mehra S, Schmid W, Spiegel R. Rapid detection of sex chromosomal aneuploidies by PCR. Indian J Med Res 1995 Mar;101:111-4.
8. Galán Gómez sindrome de Turner. Disponible en: http://www.aeped.es/protocolos/genetica/7-turner .
9. Vogel F, Motulsky AG. Human Genetics: problems and Approaches . Berlin: Sringer-Verlag; p.1982 : 48-50.
10. Nussbaum RL, McInnes RR, Willard F. Thompson & Thompson Genetics in Medicine. 6ta ed. Philadelphia: W B Saunders; 2001. 149-150.
11. Suardíaz Martínez BA, Hernánde z Pérez Y, Collazo Mesa T, Bofill Martínez AM, Gómez Martínez M, Reyes L . Diagnóstico prenatal del sexo por la técnica de reacción en cadena de la polimerasa. Rev Cubana Genet Hum 2001; 3(2). Disponible en: http://www.sigemec.sld.cu/rcgh .
12. Sybert VP, McCauley E. Turner's syndrome. N Engl J Med 2004;351(12):1227-38 .
13. Prueitt RL, Ross JL, Zinn AR. Physical mapping of nine Xq translocation breakpoints and identification of XPNPEP2 as a premature ovarian failure candidate gene. Cytogenet Cell Genet 2000;89:44-50.
14. Ogata T, Muroya M, Matsuo N. Turner syndrome and Xp deletions: clinical and molecular studies in 47 patients. J Clin Endocrinol Metab 2001;86:5498-5508.
15. On line mendelian inheritance in man. Disponible en: http://www.ncbi.nlm.nih.gov/OMIM .
16. Boucher CA, Sargent CA, Ogata T, Affara NA. Breakpoint analysis of Turner patients with partial Xp deletions: implications for the lymphoedema gene location. J Med Genet 2001;38(9):591-8.
17. Zinn AR, Ross JL. Turner syndrome and haploinsufficiency. Curr Opin Genet Dev 1998;8:322-327.
18. Haverkamp F, Wolfle J, Zerres K. Growth retardation in Turner syndrome: aneuploidy, rather than specific gene loss, may explain growth failure. J Clin Endocrinol Metab 1999;84:4578-4582.
Recibido: 15 de diciembre de 2004. Aprobado: 30 de enero de 2005.
Dra. Estela Morales Peralta. Centro Nacional de Genética Médica. 146 No. 3102, Playa, Habana 16, CP 11 600. Ciudad de La Habana, Cuba.
E-mail: estela@cngen.sld.cu
1Especialista de II Grado en Genética Clínica. Profesor Auxiliar. Centro Nacional de Genética Médica.
2Licenciado en Biología. Máster en Genética Médica. Investigador Agregado. Centro Nacional de Genética Médica
3Técnico Medio en Química. Técnico A, Centro Nacional de Genética Médica.
4Licenciado en Biología. Máster en Genética Médica. Investigador Agregado. Centro Nacional de Genética Médica.

