










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
En 1945, Adams y Oliver fueron los primeros en describir la asociación de aplasia cutis congénita (ACC) con defectos terminales transversos de extremidades.1 Desde entonces, una amplia gama fenotípica ha sido descrita2,3,4,5en estos pacientes conocido como síndrome de Adams-Oliver (SAO), que se presenta desde casos con defectos ligeros hasta defectos graves que puedan comprometer la vida del paciente.
La ACC se localiza principalmente en el vertex del cráneo y puede asociarse a diferentes grados de daño al periostio, al hueso y a la duramadre. Algunos informes describen anomalías severas del sistema nervioso central, además de alteraciones cardiovasculares, cutáneas, gastrointestinales y genitourinarias, lo cual ensombrece el pronóstico de estos pacientes.2,3,4,5 Se transmite por un patrón autosómico dominante pero también se han descrito formas autosómicas recesivas y formas esporádicas.2,6,7 Este síndrome es una entidad poco frecuente aunque existen publicaciones de algunos casos a nivel universal y apenas en el ámbito latinoamericano.8,9,10,11 En Cuba no se ha registrado pacientes afectados por este síndrome. En este trabajo se presentan dos casos con SAO, desde el punto de vista clínico, diagnosticados en la etapa neonatal, con el objetivo de aportar evidencia que demuestran la existencia de los primeros casos con este síndrome en Cuba.
PRESENTACIÓN DE CASOS
Caso 1. Recién nacido masculino de 1 día de vida, primer y único hijo de padres no consanguíneos (madre de 23 años y padre de 46 años) sin antecedentes familiares patológicos relacionados con el caso. Embarazo bajo control médico que cursa con diabetes gestacional, sin otras afecciones.
Nace de un parto distócico por cesárea a las 38,1 semanas de gestación, por la diabetes gestacional materna y cardiopatía congénita de diagnóstico prenatal del caso de estudio. Puntaje de Apgar 9/9, peso 2 700 g, talla 49 cm, circunferencia cefálica 33 cm y torácica 32 cm.
La exploración física demostró aplasia cutis y acránea parcial sobre el vértex, con afectación de ambos huesos parietales y el occipital, con encéfalo y seno venoso expuestos, cubiertos por una delgada membrana traslúcida y avascular; cutis marmorata telangiectásica generalizado, acentuado a la exposición al ambiente climatizado frío, notable prominencia de red venosa en abdomen, con mayor ingurgitación al llanto (Fig. 1).
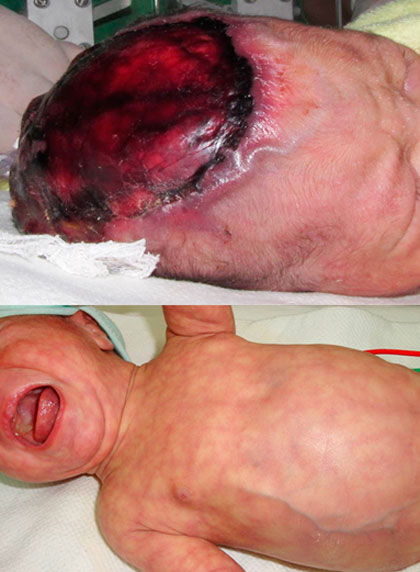
Fig. 1 Superior: aplasia cutis y acránea parcial sobre el vertex, con encéfalo y seno venoso expuestos, cubiertos por una delgada membrana traslúcida y avascular, notable prominencia de venas rodeando la lesión. Inferior: cutis marmorata telangiectásica generalizado, y notable prominencia de red venosa en abdomen.
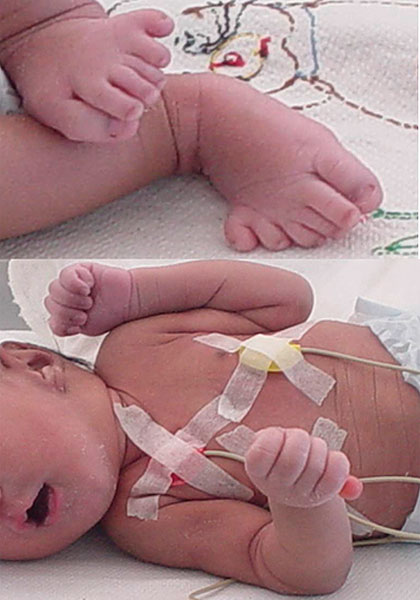
Ruidos cardiacos arrítmicos, de buena intensidad, soplo sistólico III-IV/VI en mesocardio; hipoplasia de falanges distales en pie derecho y ausencia de falange distal con pérdida de continuidad de la piel hacia el ápice del primer dedo de ese pie; ausencia de falanges distales en todos los dedos del pie izquierdo, con ausencia de piel hacia el ápice del primer dedo de ese pie. Hipoplasia de las falanges distales de todos los dedos de ambas manos con hipoplasia ungueal (Fig. 2).

Fig. 2 Superior: acortamiento de falanges distales e hipoplasia ungueal de mano izquierda. Inferior: truncaciones de las falanges distales de pie izquierdo y del primer dedo del pie derecho, con hipoplasia de falanges distales e hipoplasia ungueal de los demás dedos del pie derecho y áreas necróticas en cicatrización de ambos pies.
Ecocardiografía: asimetría de 4 cavidades con rechazo del septum interventricular hacia cavidades izquierdas. Dilatación de la aurícula derecha e implantación algo baja de la válvula tricúspide displásica (anomalía de Ebstein).
Examen oftalmológico: no encontró alteraciones.
Egresó del servicio de neonatología con seguimiento por especialistas de cardiología, neonatología, genética y neuropediatría.
Caso 2. Recién nacido femenina de 1 día de vida, cuarto hijo de padres no consanguíneos (madre de 34 años hipertensa y padre de 50 años), sin antecedentes familiares patológicos relacionados con el caso. Embarazo normal bajo control médico.
Nace de parto distócico por cesárea iterada, a las 37,4 semanas de gestación, puntaje de Apgar 3/7, con un peso de 2 700 g, talla 49,5 cm, circunferencia cefálica de 31 cm y torácica de 30 cm.
Al examen físico se encontró aplasia de piel y tejido celular subcutáneo en cráneo, en la zona inter parieto-occipital de 5 a 6 cm que deja ver la duramadre con un islote óseo y otro defecto en región occipital de 2 cm de diámetro con iguales características (Fig. 3).
En las extremidades presenta polidactilia axial en ambas manos, insertados en base del 5to. dedo, sindáctilia del 3er. y 4to. dedo del pie derecho, polidactilia en ambos pies. Pliegue simiesco en mano izquierda (Fig. 4).
Algunos rasgos dismórficos fueron implantación baja de las orejas y engrosamiento y aplanamiento de la base de la nariz.
Egresó del servicio de neonatología con seguimiento por especialistas en neonatología, genética y neuropediatría.
Por los hallazgos clínicos y los complementarios realizados se orientaron ambos casos como SAO.
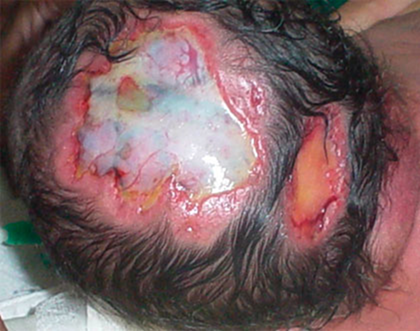
Fig. 3 Aplasia de piel y tejido celular subcutáneo en cráneo, en la zona inter parieto-occipital de 5 a 6 cm. que deja ver la duramadre con un islote óseo y otro defecto en región occipital de 2 cm de diámetro con iguales características.
DISCUSIÓN
El SAO es un trastorno descrito en 1945 por Forrest Adams y C. Peter Oliver1 que se asocia a ACC, acompañado o no de defectos del cráneo de severidad variable, con un rango muy amplio de defectos en las porciones terminales transversas de las extremidades y cutis marmorata telangiectásica congénita.4,5,6,8,12,13 Presentan un cuadro clínico fenotípico y genotípicamente heterogéneo, con un amplio espectro de manifestaciones asociadas.6 Se estima una frecuencia de 0,44/100 000 recién nacidos vivos.6
La ACC se observa en la zona posterior de la región parietal o vertex en forma de lesiones que oscilan entre 0,5 y 10 cm asociada a dilatación de venas tortuosas3,5,11 y defectos en los huesos parietales. Varias anomalías intracraneales pueden acompañar a la afectación de la ACC.4
Los defectos en las porciones terminales transversas de las extremidades son malformaciones en forma de truncaciones de las falanges distales que propician su ausencia o hipoplasias. Se han descrito casos de sindactilia (ósea o cutánea) entre 2do. y 3er. dedo, reducción de las falanges proximales y medias, polidactilia, ectrodactilia, hipoplasia de metatarsos, hipoplasia ungueal, pie varo equino, ausencia de la parte distal del miembro, micromielia y braquipodia.4,5,6,9,12,14 De esta manera, hay una amplia variabilidad en el rango de severidad, por lo que puede presentarse desde ausencia completa de pies y manos a solo ligeras manifestaciones o apariencia normal. Por lo general, están más afectadas las extremidades inferiores.15
Con cierta regularidad se hallan cardiopatías congénitas como coartación aórtica y defectos de septo ventricular y atrial, estenosis subaórtica y aórtica, estenosis de las venas pulmonares, tetralogía de Fallot, válvula mitral en paracaídas, válvula aórtica bicúspide y atresia pulmonar.3,6,13,14,16
Se ha asociado a anomalías neurológicas y del sistema nervioso central como meningitis secundaria a infecciones por medio del defecto cutáneo, encefalocele, quistes poroencefálicos, microcefalia, displasia cortical, paquigiria, hipoplasia asimétrica cerebelar, epilepsia, microcefalia, hipoplasias del cuerpo calloso, calcificaciones intracraneales del tipo de las infecciones TORCH, displasia de la corteza cerebral e hidrocefalia y retraso psicomotor.5,6,14
Otras anomalías poco comunes incluyen hipoplasia del nervio óptico, microftalmia, alteraciones oculares, malformación del vítreo y la retina, cataratas congénitas bilaterales, defectos en los pabellones auditivos, apéndices cutáneos de los dedos de los pies, ACC de la rodilla, hiperpigmentaciones focales, hemangiomas, malformaciones arteriovenosas del cuero cabelludo, criptorquidia, politelia, retraso del crecimiento, déficit mental, baja talla, esclerosis hepatoportal, anomalías intrahepáticas, fibrosis portales, dolicocefalia y paladar hendido, aplanamiento de la base de la nariz4,5,8,12,17
Snape4 y otros, sugieren el uso de criterios mayores y menores para ayudar a alcanzar el diagnóstico de SAO. La ACC, las anomalías transversas distales de las extremidades y el antecedente familiar de SAO son considerados como signos mayores para el diagnóstico. El cutis marmorata telangiectásico congénito, los defectos cardiovasculares y el resto de las manifestaciones son considerados signos menores. La presencia de dos signos mayores es suficiente para hacer el diagnóstico. La combinación de un criterio mayor y uno menor coloca al paciente en un alto índice de sospecha.4
Nuestros pacientes, ambos, presentaron ACC y anomalías transversas distales de las extremidades como signos mayores. El caso 1 presentaba además cutis marmorata telangiectásica y cardiopatía congénita como signos menores. La cardiopatía diagnosticada por ecocardiografía fue una anomalía de Ebstein, no publicada en la literatura revisada, aunque sí se ha señalado atresia o incompetencia de la válvula tricúspide.5
La mayoría de los casos tienen un patrón de herencia autosómica dominante con expresión variable intra- e interfamiliar, recientemente asociada con la mutación del gen ARHGAP31 localizado en 3q13 resultando en pérdida de la actividad del CDC42 con disrupción de la estructura de actina del citoesqueleto celular, y con la mutación del gen RBPJ en 4p15. También ha sido demostrada la herencia autosómica recesiva, además, la ocurrencia de casos esporádicos. Se señala que no hay diferencias en la expresión clínica de esta afección para cualquiera de los patrones de herencia referidos. Se han identificado seis genes causales: ARHGAP31 ya mencionado, DOCK6, EOGT, RBPJ, NOTCH1, and DLL4.5,18,19,20
En nuestros pacientes no se recogieron antecedentes familiares de esta enfermedad, ni hay consanguinidad, por lo que planteamos que se tratan de casos de presentación esporádicos. Varios autores relacionan la edad avanzada del padre con la presentación de este síndrome6 y en estos dos casos los padres, aunque no pueden conceptualizarse como de edad avanzada, llama la atención la diferencia de edades, pues ambos tenían más de 15 años concernientes a las respectivas madres.
La patogenia es desconocida. Inicialmente Adams y Oliver1 sugirieron que se debía a detención del desarrollo o ausencias de ciertas partes del esqueleto y tejidos blandos. Actualmente la teoría vascular es la más aceptada, en la que se plantea una predisposición intrínseca a la interrupción del flujo sanguíneo de los pequeños vasos durante la embriogénesis. Esta anomalía vascular pudiera ser el resultado de un defecto genético que cause disminución de la estabilidad de los vasos sanguíneos embrionarios hacia las fuerzas tensoras en el período de 6 a 8 semanas de vida embrionaria.6 La presencia de cutis marmorata y de venas dilatadas y tortuosas en el vertex son considerados indicadores de una predisposición a un compromiso vascular en ciertas áreas como el vértex y los miembros.6
La presentación de un amplio espectro de manifestaciones clínicas implica la recomendación de una atención multidisciplinaria.
Debido a lo expuesto anteriormente el pronóstico es muy variable y va a depender fundamentalmente del grado de afectación del sistema nervioso central y el sistema cardiovascular.
En conclusiones podemos decir que estamos en presencia de dos pacientes con SAO, ambos de presentación esporádica y uno de estos con una cardiopatía congénita (anomalía de Ebstein) no informada hasta ahora en la literatura.

