










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkLa hemocromatosis hereditaria es el defecto genético más frecuente en personas de origen caucásico. En el año 1935 ya se conocía que esta afección se caracteriza por depósitos de hierro aumentados en los tejidos; en la década del 70 se reconoció como un trastorno autosómico recesivo ligado a la región del brazo corto del cromosoma 6 que codifica para el HLA-A*3. En el año 1996 fue finalmente identificado el gen HFE como el responsable de la enfermedad.1
La mutación genética consiste en la sustitución de tirosina por cisteína en la posición 282 de la proteína HFE (282Y). Fue originada por casualidad en ancestros celtas (o vikingos) en Europa noroccidental hace 2000 años, no provoca serios obstáculos para la reproducción, y confiere algunas ventajas como: la resistencia a la deficiencia de hierro dietético y ciertas infecciones.2 La homocigozidad para la mutación C282Y existe en 5 de cada 1 000 personas descendientes de Europa del norte, o sea, se constata una prevalencia diez veces mayor que la de los genotipos de la fibrosis quística. Esto provoca una predisposición genética a una cadena de sucesos que culmina en daño tisular grave,3 aunque por razones desconocidas solo menos de un 10 % de estas personas desarrollarán manifestaciones clínicas de hemocromatosis.4
El daño orgánico sintomático generalmente aparece en la cuarta década de la vida, con síntomas no específicos como: fatiga inexplicada y artralgias. La enfermedad hepática puede cursar con hipertransaminasemia discreta y asintomática, o con la presencia de: cirrosis, insuficiencia hepática e incluso carcinoma hepatocelular. También aparecen endocrinopatías (diabetes, hipogonadismo, hipogonadotrópo, impotencia e hipotiroidismo), cardiopatías (arritmias e insuficiencia cardíaca) y enfermedad articular (artritis destructiva).5,6
El diagnóstico se realiza a través de los marcadores indirectos para los depósitos de hierro (saturación de transferrina y ferritina), y el estudio genético puede mostrar los genotipos C282Y/C282Y o C282Y/H63D.1,7,8
En la actualidad, se recomienda la biopsia de hígado para definir en qué estadio está la enfermedad hepática (en homocigotos C282Y o en heterocigotos C282Y/H63D, si se asocia en estos últimos a hipertransaminasemia o a niveles de ferritina mayores de 1000 µg/ml).9,10, A pesar de otras terapias novedosas (transferrina y hepcidina sintética, o análogos y agonistas de hepcidina) en fase investigativa, la flebotomía constituye la piedra angular del tratamiento para reducir los niveles plasmáticos y tisulares de hierro. Generalmente es bien tolerada y sin riesgos significativos de anemia para el enfermo.8 Esta condición clínica resulta poco común en la práctica médica, y puede confundirse fácilmente con otras enfermedades dada la presencia de signos y síntomas inespecíficos. Esta cuestión requiere de un alto índice de sospecha para indicar las investigaciones pertinentes y lograr un diagnóstico acertado. Por tales motivos, se considera oportuno presentar este paciente con diagnóstico de hemocromatosis hereditaria.
Presentación del paciente
Paciente masculino de 49 años de edad con antecedentes de aparente salud hasta hace alrededor de dos años, en los que comenzó a experimentar astenia, anorexia y artralgias. El paciente fue valorado por especialistas de Gastroenterología; se constató hipertransaminasemia y sero-negatividad para virus B y C. Se interpretó esta situación como presunta hepatitis reactiva a giardiasis. Transcurridos cinco meses fue evaluado otra vez con los síntomas y signos ya descritos; resultó llamativa la hiperpigmentación cutánea en áreas del tronco y extremidades superiores (Figura 1) además de dolor a la palpación en hipocondrio derecho.
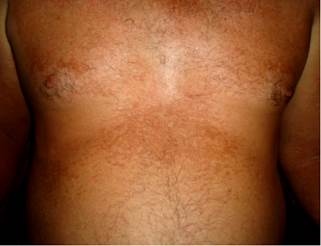
Figura 1 Se puede observar la hiperpigmentación cutánea en áreas del tronco y extremidades superiores del paciente
No se encontró anemia ni aceleración marcada de la velocidad de sedimentación globular, aunque los niveles de ferritina (1395 ng/ml), saturación de transferrina (79 %) y hierro (48 µmol/l) si estaban francamente elevados. Los niveles de albúmina y bilirrubina y los tiempos de protrombina y parcial de trombo-plastina estaban dentro del rango normal, y el estudio genético, indicado posteriormente, mostró un genotipo C282Y/C282Y.
Ante la fuerte sospecha de hemocromatosis el paciente fue remitido al Instituto de Gastroenterología, para realizarle una biopsia de hígado y definir en qué estadio estaba la enfermedad hepática; se consideró la presencia de hipertransaminasemia, niveles de ferritina superiores a 1000 µg/ml y los resultados del estudio genético. Los resultados histológicos del estudio de la biopsia fueron compatibles con el daño tisular propio de la enfermedad sospechada: signos de cirrosis con extensos depósitos férricos (Figuras 2 y 3).
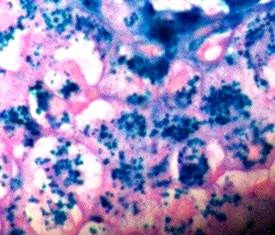
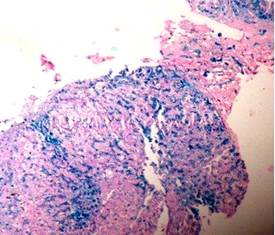
La ultrasonografía Doppler de hígado mostró signos de hipertensión portal incipiente, y los estudios de ferrocinética revelaron hallazgos compatibles con hemocromatosis.
El paciente se mantuvo clínicamente estable, y seis meses después de haber iniciado las flebotomías (500 ml una vez por semana) los síntomas mejoraron. El régimen terapéutico se mantuvo durante diez meses, después de este período el paciente declinó continuarlo. También los niveles de ferritina y saturación de transferrina descendieron. En el momento de presentar el caso los síntomas no habían empeorado, y no se habían detectado signos clínicos de insuficiencia cardíaca, hipotiroidismo u otras complicaciones.
Comentario
Las manifestaciones clínicas y las alteraciones encontradas en los estudios diagnósticos realizados coincidieron con otros casos descritos en la literatura médica; sin embargo, a pesar de la alta incidencia de la homocigozidad del C282Y en individuos de origen caucásico, es infrecuente la presencia de todos los signos y síntomas clásicos de esta condición clínica.1,3
Los cuatro principales mecanismos fisiopatológicos son: la absorción incrementada de hierro dietético en el tracto digestivo alto, la expresión disminuida de la hormona reguladora de hierro (hepcidina), la cual también contribuye al aumento en la absorción intestinal de hierro, trastornos en la función de la proteína HFE, el daño tisular y la fibrogénesis inducida por el hierro.6
La ausencia de anemia con evidencias de sobrecarga férrica conllevó a que otras condiciones fueran descartadas: la anemia sideroblástica, la anemia hemolítica crónica y la talasemia mayor. La presencia de hipertransaminasemia y la negatividad para los marcadores virales de la infección por virus B y C descartaron la hepatitis aguda o crónica. Los hallazgos histológicos excluyeron la posibilidad de otras condiciones como la esteatohepatitis no alcohólica. Es importante señalar que menos del 10% de los pacientes homocigotos para C282Y desarrollan sobrecarga férrica grave, con daño orgánico y manifestaciones clínicas (esto corresponde al estadio 3 o final).7) Aun cuando en el medio existe controversia con el uso de la flebotomía, se inició un régimen con frecuencia semanal; se consideró que esta es la piedra angular del tratamiento aun en presencia de complicaciones como la cirrosis. Por lo general, debe mantenerse en la mayoría de los casos dado el riesgo de carcinoma hepatocelular y para garantizar la efectividad del trasplante hepático (es bien tolerada incluso cuando se realiza de forma intensiva). Se ha demostrado que su uso prolonga la supervivencia pues hay informes sobre la regresión de la fibrosis hepática en un 13-50 % de los casos. El uso de quelantes de hierro solo se justifica en presencia de anemia u otras situaciones.1,2,3 Otros nuevos agentes quelantes se encuentran en fase investigativa, sucede igual con algunas terapias novedosas como: la transferrina y hepcidina sintéticas; otros análogos y agonistas de la hepcidina también podrían ser efectivos.8
Se recomienda que en todo paciente con síntomas y signos sugestivos, o con historia familiar en parientes de primer grado, se investigue la presencia de sobrecarga férrica a través de la saturación de transferrina y ferritina. Es oportuno realizar un pesquisaje genético de la mutación antes mencionada, para lograr un diagnóstico temprano y la prevención de futuras complicaciones.

