










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El síndrome de Currarino es una enfermedad hereditaria poco frecuente, constituida por una tríada compuesta por malformación ano rectal, masa presacra y alteraciones sacras. Por primera vez se reporta esta asociación por Kennedy en 1926 pero no es hasta 1981 que Guido Currarino la describe. 1
El patrón de herencia es autosómico dominante; sin embargo, se presentan casos esporádicos asociada a una mutación del gen HLXB9 localizado en el cromosoma 7, en la región 7q36. 2,3
Este síndrome pertenece al grupo de malformaciones neuroentéricas, estando asociado a la constipación crónica. Currarino y cols. propusieron que adherencias neuroectodérmicas y defectos en la notocorda durante la vida fetal temprana pueden resultar en una fístula entre el intestino y el canal espinal con presencia de elementos entéricos ventralmente y neurales dorsalmente. 4) Aunque la incidencia real de este raro síndrome se desconoce, existen menos de 300 casos publicados en la literatura mundial, pero se cree que muchos pasan desapercibidos sin ser diagnosticados, debido principalmente a la variabilidad fenotípica intra e interfamiliar de este síndrome. 3,4
El diagnóstico prenatal del síndrome de Currarino es importante para poder evitar complicaciones derivadas de la masa presacra y para dirigir el tratamiento. A continuación se describe un caso de una fetal intermedia con un síndrome de Currarino cuyo valor científico radica en lo infrecuente de su diagnóstico en etapa fetal pues generalmente son diagnosticados en etapa posnatal.
CASO CLÍNICO
Gestante de 30 años de edad, con antecedente de salud aparente, con historia obstétrica de presentar una gestación, sin haber antecedidos partos y abortos, atendida en la consulta de control prenatal sin complicaciones previas, quien inició control de embarazo a partir de la semana 12, con estudios de laboratorio prenatales dentro de parámetros normales. En el ultrasonido estructural a la semana 22 de gestación se le detectó al feto una malformación renal y oligoamnio. Debido a los resultados en la ecografía, la paciente se remitió, evolucionó y valoró por el Servicio de Genética Nacional, donde tras estudios de imágenes se constataron los mismos hallazgos y otros como defecto sacroccígeo y masa presacra, los cuales coinciden con el diagnóstico de síndrome Currarino.
Teniendo en cuenta los riesgos que supone este síndrome para el feto debido a la aparición de compromiso neurológico importante, disfunción vesical e intestinal severa se informó a los padres sobre el estado y pronóstico fetal, además las posibles secuelas derivadas del defecto observado y la pertinencia de la interrupción del embarazo; ellos manifestaron estar de acuerdo, asimismo lo expresaron por escrito y firmaron su consentimiento.
La interrupción del embarazo se realizó por vía vaginal, en la interrupción se observó un feto de sexo masculino, con 625 g de peso, sin actividad cardíaca, en el que se observaron malformaciones a nivel de la región sacra; se le realizó la necropsia y en el examen macroscópico se describieron los siguientes hallazgos:
Defecto sacro coccígeo específicamente una ausencia total del sacro.
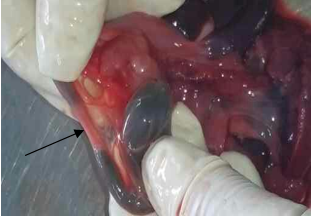
Masa o tumoración presacra (de aspecto quístico). (Foto 1)
Ano imperforado y ausencia de pliegue interglúteo.
Bolsa escrotal única. (Foto 2a)
Agenesia del sacro. (Foto 2b)
Estenosis del sigmoide (Foto 3)
Riñón único, pélvico y poliquístico, con salida de 2 uréteres.


En el examen histológico se halló:
Masa presacra: se correspondió con un mielomeningocele.
Riñón poliquístico.
Patrón pulmonar en fase canalicular e inmadurez generalizada propia de la edad gestacional.
Como diagnóstico patológico definitivo se planteó un síndrome de Currarino diagnosticado en edad fetal.
DISCUSIÓN
Se presentó el caso del diagnóstico prenatal de un síndrome de Currarino completo, con presencia de ausencia total del sacro, estenosis del sigmoide, ano imperforado y una tumoración presacra que se correspondió con un mielomeningocele.
El Síndrome de Currarino es una patología poco frecuente. Está caracterizado por tres signos clínicos claves: estenosis anal, malformaciones sacro coccígeas y masa presacra. Su prevalencia es desconocida. Rara vez el diagnóstico se realiza durante el período prenatal ya que suele manifestarse con mayor frecuencia en la infancia. 5
Currarino postuló que las alteraciones características de la tríada podrían ser causadas por adherencias entre el endodermo (que da origen al intestino posterior) y el ectodermo (que da origen al tubo neural). La persistencia de estas adherencias antes de la aparición de la notocorda causaría que ésta se divida o se desplace, impidiendo la fusión anterior de las vértebras. También, propuso que podría tratarse de una separación primaria de la notocorda causando adherencias entre el endodermo y el ectodermo, con el secundariodesarrollo vertebral anormal. La reabsorción parcial de la fístula causaría alteraciones como mielomeningocele anterior o quiste entérico. La malformación ano rectal sería explicada por la presencia de estas adherencias entre el intestino posterior y el tubo neural. 6
El defecto óseo del sacro es el componente principal del síndrome que está presente en todos los casos y corresponde a la malformación sacra tipo IV o sacro “en cimitarra” donde se observa presencia de un hemisacro con preservación en la morfología de S1. El “sacro en cimitarra” es el hallazgo patognomónico del síndrome de Currarino.
El tipo de malformación anorrectal más frecuentemente encontrado en el síndrome de Currarino es la fístula recto perineal, sin embargo, diferentes tipos de malformación anorrectales (fístula recto uretral, fístula recto vestibular o recto espinal) pueden estar presentes.
La masa presacra puede ser un meningocele anterior, un teratoma (generalmente benigno cuando se presenta en el contexto de este síndrome), un quiste dermoide, una duplicación rectal u otro tumor no común.
Adicionalmente, puede haber otras malformacionesasociadas como: malformaciones urológicas (dobleuréter, doble riñón, reflujo vesicoureteral, riñón displásicoohipospadias), malformaciones genitales (úterobicorne, vagina septada, genitales ambiguos y clítorisbífido) y malformaciones en el sistema nervioso central (malformación de Arnold Chiari tipo I). 7
La expresividad del síndrome es muy variable, siendo posible que la tríada se manifieste de forma completa o incompleta, desde la alteración aislada del sacro hasta el espectro completo de la enfermedad. Se propone una clasificación de acuerdo con la presencia de las alteraciones y características:
Síndrome de Currarino completo cuando existe la presencia de hemisacro, malformación anorrectal y masa presacra. En estos casos el diagnóstico temprano es posible incluso desde el nacimiento.
Síndrome de Currarino incompleto cuando está presente el hemisacro y una de las otras alteraciones. En estos casos, si está presente la malformación anorrectal el diagnóstico es posible desde el nacimiento, mientras que si es la masa presacra la que está presente, el diagnóstico es más difícil y la presentación puede ser a edades más tardías.
Síndrome de Currarino mínimo cuando solo el hemisacro está presente. En estos casos su presentación y diagnóstico suelen ser más tardíos. 7
CONCLUSIONES
El síndrome de Currarino es poco frecuente y puede manifestarse con una tríada de malformaciones. El análisis del caso y la revisión de la literatura permiten ilustrar a la comunidad científica que mediante un control obstétrico adecuado en edad gestacional temprana y un tamizaje correcto de las malformaciones fetales se puede garantizar un diagnóstico oportuno de este síndrome.

