










 Servicios personalizados
Servicios personalizados Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La seguridad y eficacia de los productos farmacéuticos están influenciados por sus propiedades intrínsecas y por su estabilidad, la que depende de factores ambientales y de los relacionados con el producto, como las propiedades físico-químicas del ingrediente activo y de los excipientes, forma farmacéutica, formulación, procesos de fabricación y envase. Un ensayo de estabilidad determina los parámetros de calidad del producto en condiciones de almacenamiento específicas, durante un tiempo que avale su fecha de vencimiento (Centro para el Control Estatal de Medicamentos, Equipos y Dispositivos Médicos (CECMED, 2000); (International Conference of Harmonization ICH, 1998).
Ensayos de estabilidad a largo plazo del Policosanol en sus formas terminadas (PPG 5, 10 y 20 mg) han demostrado que el tiempo de vida útil de estos productos es 60 meses, según consta en sus Registros Sanitarios (CECMED, 2017a; CECMED, 2017b; CECMED, 2017c). Dichos ensayos fueron realizados durante la Investigación-Desarrollo (I+D) de esas tres formulaciones, según Regulación aprobada (CECMED, 2000), la cual no exige que la estabilidad, una vez registrado el producto, vuelva a ser monitoreada en un programa continuo. Sin embargo, de acuerdo con las Directrices de Buenas Prácticas de Fabricación (GMP, por sus siglas en inglés) de la Unión Europea, que entraron en vigor en 2006 (EU, s.f.), se comenzaron a exigir, como parte de la Revisión Anual de Calidad de todo medicamento registrado, un control de su estabilidad en curso (“ongoing”), con vistas a demostrar que este mantiene su calidad de forma estable durante toda su vida comercial. En consonancia con lo anterior, el CECMED aprobó en 2012 una resolución que exige la realización de estudios “ongoing” para los productos registrados (CECMED, 2012).
La inclusión de estudios de estabilidad en la Revisión Anual de Calidad del producto, ha conllevado a que áreas de control de calidad, que formaban parte de I+D, ahora se incluyan en las inspecciones oficiales de GMP. Este es el caso, por ejemplo, de la cualificación de los equipos y laboratorios implicados en los ensayos de estabilidad. Además, se exige la implementación de procedimientos para tratar los resultados fuera de especificación, no solo para liberar los lotes sino también durante las pruebas de estabilidad, y para evaluar los resultados fuera de tendencia. Consecuentemente, los ensayos se deben de realizar de acuerdo con los requisitos de GMP, teniendo en cuenta que la estabilidad en curso difiere de otras pruebas de estabilidad (Tabla 1) (ICH, 1999; ICH, 2002).
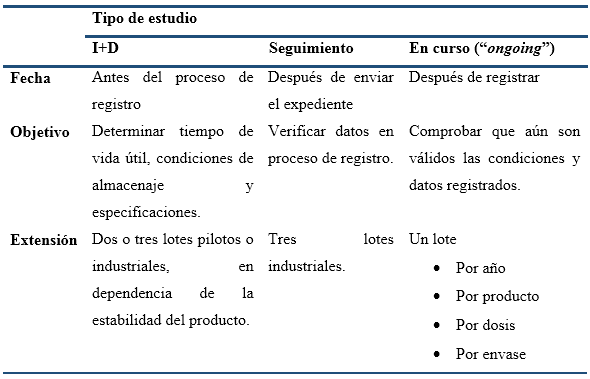
Los ensayos de estabilidad en curso deben aplicarse a todos los medicamentos, en cada dosificación y tipo de envase primario, incluyendo como mínimo un lote al año en condiciones a largo plazo, de forma continua durante todo el período de vida útil etiquetado. Durante dicho período, se demostrará si, “en condiciones reales”, el producto conserva la calidad definida en su documento de registro.
Los ensayos de estabilidad realizados para el registro solo proporcionan una "instantánea". Los posibles efectos adversos, por cambios en la fabricación y la cadena de suministro, deben identificarse y evaluarse mediante la estabilidad en curso, cuyos protocolos pueden reducir la frecuencia de las pruebas y no evaluar parámetros que no sean críticos (Tabla 2). En este sentido, los factores críticos son dosis y contenido de impurezas, y los menos críticos los organolépticos, físicos, desintegración y uniformidad. No obstante, al final de la vida útil, todos los parámetros de prueba deben verificarse nuevamente, para demostrar la estabilidad durante todo ese período (ICH, 2002).
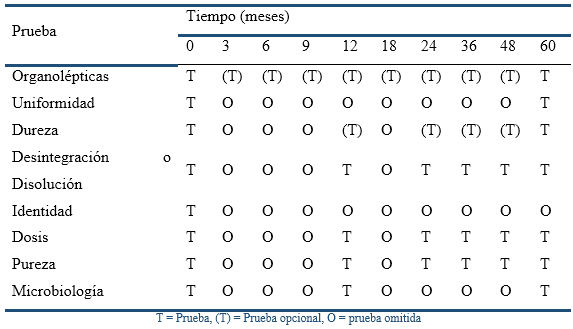
Por otra parte, si el producto se fabrica en diferentes dosis con matrices similares y en diferentes tamaños de envase, se pueden aplicar las recomendaciones de “bracketing” (ICH, 2000). Por ejemplo, si hay más de dos tamaños de envase se permite ensayar solo los extremos, y el efecto de diferentes envases secundarios pudiera obviarse.
Los resultados de las pruebas de estabilidad en curso deben compararse con los de pruebas anteriores, pueden utilizarse en el análisis de tendencias, y deben ser incorporados y discutidos en la Revisión Anual del Producto. En el caso de resultados fuera de especificación o tendencias negativas, se debe comunicar a las autoridades de control, y además se deben revisar las áreas críticas de producción, para identificar la causa y evitar la transmisión a otros lotes o productos; así como revisar de forma crítica la estabilidad etiquetada, retirar el lote afectado, así como implementar medidas correctivas.
Teniendo en cuenta lo abordado anteriormente, el objetivo del presente estudio fue conocer la estabilidad de lotes industriales de tabletas PPG 5, 10 y 20, un lote por año, con vistas a comprobar la estabilidad de estas formas terminadas durante todo el tiempo de vida útil aprobado (60 meses), en condiciones ambientales de la zona climática IV.
MATERIALES Y MÉTODOS
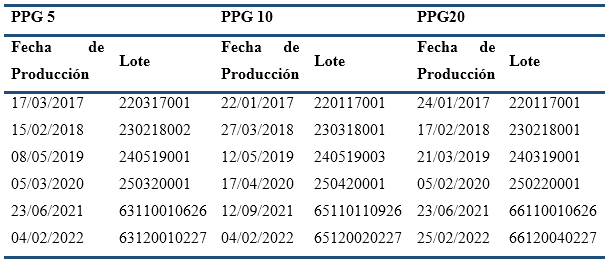
Diseño General del estudio. Los ensayos se llevaron a cabo a 30 ( 2(C y 70 ( 5% de humedad relativa, teniendo en cuenta las Regulaciones 23 y 61 del CECMED (CECMED, 2000; CECMED, 2012), así como las directrices internacionales (EU, s.f; ICH, 1999; ICH, 2000; ICH, 2002). Se utilizaron tabletas revestidas de Policosanol, con 5, 10 y 20 mg de alcoholes grasos, envasadas en blísteres de cloruro de polivinilo/aluminio, producidas industrialmente (SolMed, Cuba). Hasta el presente se han incluido en el ensayo muestras de un lote anual de cada formulación desde 2017 (Tabla 3), y posteriormente, cada año, se incorporará un nuevo lote de cada formulación y saldrán del estudio los lotes que cumplan 60 meses de almacenaje. Los muestreos se efectuaron anualmente, tomando como tiempo cero la fecha de liberación de cada lote.
Tabla 3 Lotes de tabletas de Policosanol 5, 10 y 20 mg incluidos en el ensayo de estabilidad en curso.
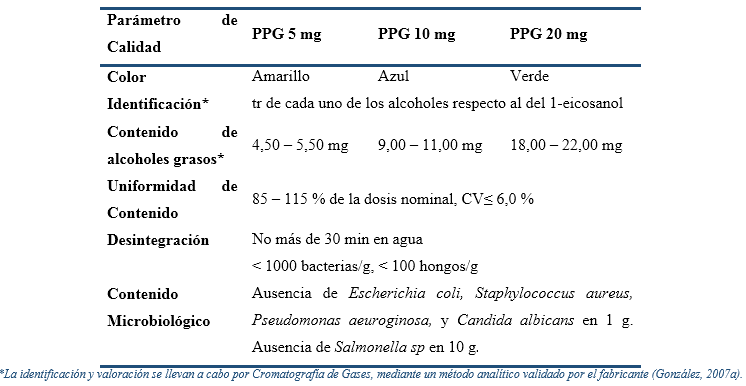
Los controles se realizaron por métodos oficiales de Farmacopea (United Stated Pharmacopoeia USP, 2017) o por métodos del fabricante (González, 2007a), según lo declarado en los Registros Sanitarios respectivos (CECMED, 2017a; CECMED, 2017b; CECMED, 2017c), y las determinaciones realizadas en cada muestreo estuvieron en dependencia de cuán crítico de consideró cada parámetro. En este sentido, entre 12 y 48 meses solo se midieron las características organolépticas, el tiempo de desintegración, el contenido de alcoholes y la presencia de productos de degradación, mientras que al cumplirse el tiempo de vida útil (60 meses), se evaluaron todos los parámetros de calidad, por lo que se determinaron además la uniformidad de contenido y el contenido microbiológico. Se aceptaron resultados dentro de los límites definidos en las especificaciones de calidad de cada producto (Tabla 4).
Equipos. Balanza analítica, termostato seco, desintegrador de tabletas, generador de hidrógeno, termohigrómetro digital y cromatógrafo de gases CG-14B con detector de ionización por llama, sistema de cómputo (Shimadzu, Japón) y una columna BPX - 5 (30 m x 0,53 mm di y 1,5 (m de espesor de película) de 220 a 320°C a 8 0C/min, con 15 min a la temperatura final; el detector y el inyector a 320 °C, con hidrógeno como gas portador a 14 mL/min y un volumen de inyección de 1 (L.
Reactivos. Cloroformo como disolvente, N-metil, N-trimetilsilil trifluoroacetamida p.a. (Sigma, EUA) como agente derivatizante; 1- tetracosanol (C24), 1-hexacosanol (C26), 1-heptacosanol (C27), 1-octacosanol (C28), 1-triacontanol (C30) (Sigma, USA, 99%) como sustancias de referencia y 1 - eicosanol (C20) como patrón interno.
RESULTADOS Y DISCUSIÓN
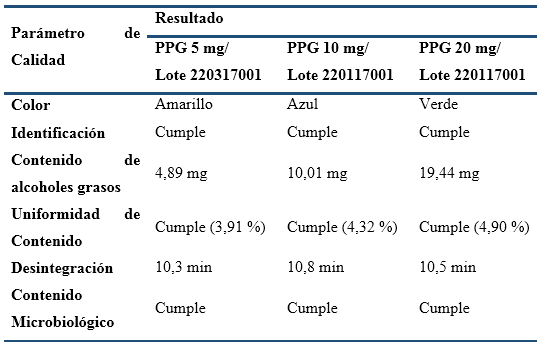
En la Tabla 5 se presentan los resultados de los parámetros de calidad determinados a las muestras con 60 meses en la Zona Climática IV, siendo estos los primeros lotes del ensayo que cumplen con este tiempo de almacenaje. Como se puede apreciar, los tres lotes cumplieron con todas sus Especificaciones de Calidad hasta el tiempo de vida útil declarado, lo cual permite corroborar las altas estabilidades de estas formulaciones, determinadas anteriormente mediante estudios de I + D previos a la aprobación de sus Registros. Los demás resultados del ensayo, con las tres formulaciones de Policosanol (muestreos a 12, 24, 36 y 48 meses de almacenaje), tampoco mostraron cambios en los parámetros evaluados, todos los cuales se mantuvieron cumpliendo con las especificaciones de calidad.
Tabla 5 Resultados del ensayo de estabilidad en curso, Tabletas de PPG 5, 10 y 20 mg, con 60 meses de almacenaje en las condiciones de la Zona Climática IV.
En ningún caso se detectaron tampoco señales cromatográficas que evidenciaran la aparición de productos de degradación (contenidos inferiores a 0,01 mg), al ser analizadas las muestras según métodos cromatográficos descritos previamente (Sierra, 2005; González, 2007b). En las Figuras 1, 2 y 3 se pueden observar cromatogramas característicos de las 3 formulaciones, en el muestreo correspondiente a los 60 meses. En todas estas figuras, correspondientes a los muestreos con mayor tiempo de almacenaje, se aprecia que no aparecieron productos de degradación (zona del cromatograma posterior a los 20 min), lo cual concuerda con que no se apreciara disminución en el contenido de los alcoholes grasos en ningún caso durante todo el estudio.
Al no haber disminuido el contenido de alcoholes grasos en ninguno de los lotes, lo cual se aprecia por simple inspección de las tablas, y queda corroborado por la ausencia de productos de degradación, no se justifica la realización de pruebas estadísticas. Teniendo en cuenta estos resultados, podemos afirmar que todos los lotes evaluados, de las tres dosis, fueron estables durante todo el tiempo de almacenaje al que se han sometido hasta el presente (60 meses). Muestreos posteriores permitirán seguir comprobando el cumplimiento del tiempo de vida útil aprobado para estas formulaciones.



CONCLUSIONES
Las tres formulaciones de tabletas de Policosanol (con dosis de 5, 10 y 20 mg de alcoholes grasos), evaluadas mediante ensayos de estabilidad en curso, cumplieron con los parámetros de calidad determinados en todos los muestreos realizados hasta los 60 meses de almacenaje en las condiciones de la Zona Climática IV, sin mostrar evidencias de degradación, lo cual demuestra el cumplimiento del tiempo de vida útil que consta en los Registros Sanitarios de estas formulaciones.

