










 Servicios personalizados
Servicios personalizados Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Los avances actuales de la industria cosmetológica, han propiciado el desarrollo de una amplia variedad de productos dermocosméticos para el cuidado profundo, continuado y eficaz de la piel, entre los que se encuentran nuevas cremas y jabones que contribuyen en gran medida a la limpieza y nutrición cutánea, así como a su revitalización desde las capas más profundas (Minero-Diaz, 2017).
Los alcoholes grasos 1-octacosanol (C28) y 1-tetracosanol (C30), así como los extractos de ceras naturales donde estos alcoholes aparecen en altas proporciones, han demostrado efectos antiinflamatorios y antioxidantes, al actuar sobre enzimas involucradas en dichos procesos (Pérez et al., 2013; 2014; 2015; Ravelo et al., 2010; 2011; 2013; 2016; Pérez et al., 2015). El 1-octacosanol inhibe marcadamente la actividad de ciclooxigenasa 1, sin afectar la ciclooxigenasa 2 ni la 5-lipooxigenasa, mientras que el 1-triacontanol inhibe marcadamente la 5-lipooxigenasa y moderadamente la ciclooxigenasa 2, sin cambiar la actividad de la ciclooxigenasa 1 (Ravelo et al., 2016). Como parte de los procesos de obtención de los ingredientes farmacéuticos activos D-001 y D-002, en el Centro Nacional de Investigaciones Científicas se obtienen subproductos ricos en alcoholes grasos, principalmente en 1-octacosanol y 1-triacontanol, los que fueron utilizados en el desarrollo de una crema para uso tópico, con efectos antiinflamatorios y antioxidantes.
Las regulaciones actuales exigen que las metodologías analíticas desarrolladas para determinar los ingredientes farmacéuticos activos, en cualquier forma terminada, estén debidamente validadas, de forma tal que permitan garantizar un alto grado de confianza y seguridad en sus resultados (Centro para el Control Estatal de la Calidad de los Medicamentos CEDMED, 2014; International Conference of Harmonization. ICH, 2000). El desarrollo de esta crema requirió la puesta a punto y validación de una metodología analítica para determinar su dosis, la cual será empleada como parte de su control de calidad y en sus estudios de estabilidad.
MATERIALES Y MÉTODOS
Reactivos y Disoluciones
Se empleó el lote de crema 0116 (Centro Nacional de Investigaciones Científicas, Cuba), N-metil, N-trimetilsililtrifluoroacetamida (MSTFA) como agente derivatizante, 1-eicosanol (C20), 1-octacosanol (C28) y 1-triacontanol (C30) como sustancias de referencia, peróxido de hidrógeno al 30 %, metanol y cloroformo, de calidad analítica (Sigma, EUA). Además, se prepararon las disoluciones siguientes:
Disolución de patrón interno (DPI): 1-eicosanol (0,4 mg/mL) en cloroformo.
Disolución de referencia de alcoholes (DRA): C28 (0,50 mg/mL) y C30 (0,50 mg/mL) en cloroformo.
Disolución matriz de referencia de alcoholes (DMR): 0,5 mL de DRA se llevaron a sequedad, se añadieron 0,150 mL de DPI y 0,05 mL de MSTFA; se cerró el vial y se calentó a 65 °C durante 15 min.
Disolución de ácido clorhídrico (HCl) 0,1 mol/L.
Disolución de hidróxido de sodio (NaOH) 0,1 mol/L
Equipos
Se empleó un cromatógrafo de gases GC-14A con detector de ionización por llama (Shimadzu, Japón), equipado con una columna BPX-5 (30 m x 0,53 mm d.i. x 1,5 μm de espesor de película, SGE, Australia) con un programa de 180°C (1 min) hasta 320 °C (5 min) a 8 °C/min. El detector y el inyector se mantuvieron a 320 °C, el volumen de inyección fue 1 μL en modo splitless, y el flujo del gas portador (hidrógeno) fue 8 mL/min. Para la formación de la llama se utilizaron flujos de 40 y 400 mL/min, para el hidrógeno y aire, respectivamente. Se emplearon además una balanza analítica, zaranda, centrífuga, termostato seco, generador de hidrógeno y compresor de aire.
Preparación de la muestra
Se pesó 1 g de crema en un vial de 20 ml (n = 3), se le añadieron 3 ml de una mezcla metanol:agua (80:20 v/v) y 5 ml de la DPI. Se agitó en zaranda durante 30 min, se centrifugó durante 5 min a 1500 rpm, se extrajo la fase orgánica hacia un vial de 20 ml, se evaporó hasta sequedad a 65°C con ayuda de corriente de aire y se adicionaron 2 mL de cloroformo. Se calentó a 65°C durante 3 min y se transfirió una alícuota de 500 μl hacia un tubo de ensayos, donde se añadieron 60 μl de MSTFA y se calentó a 65°C durante 15 min para su análisis por cromatografía de gases.
Método analítico
La determinación cualitativa se basó en la comparación de las retenciones relativas, con las obtenidas al analizar, en iguales condiciones, muestras de referencia de los alcoholes C28 y C30, empleando al alcohol C20 como patrón interno. La cuantificación se realizó por el método del patrón interno, previo cálculo de los factores másicos de respuesta relativa, con el empleo de la DMR, según los métodos de cálculo anteriormente descritos por González, et al. (2006).
Validación
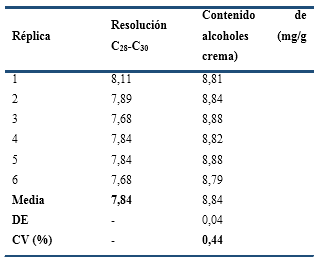
La Aplicabilidad del Sistema se determinó a partir de seis inyecciones de una misma réplica, preparada según la metodología descrita anteriormente. A partir de los cromatogramas obtenidos se determinó la resolución cromatográfica entre los alcoholes C28 y C30 (la cual debía ser mayor de 2,5) y la repetibilidad de la inyección, dada por el coeficiente de variación (CV %) con que se cuantificó el total de los alcoholes, cuyo límite de aceptación fue < 0,5 % (ICH, 2000; CECMED, 2014).
La Linealidad del Método constó de 5 puntos (entre 50 y 150 % de la concentración nominal de alcoholes en la crema) con 3 réplicas en cada punto. Para este ensayo la crema se preparó con los alcoholes 1-octacosanol y 1-triacontanol, sustancias de referencia (Sigma, USA), teniendo en cuenta que, como promedio, el contenido de estos alcoholes grasos en la crema se encuentra entre 8 y 10 mg/g de crema. El cálculo de la ecuación de regresión (y = bx +a) y del coeficiente de correlación (r) se hizo por mínimos cuadrados (Microsoft Excel). Para ello se analizaron las relaciones de las áreas obtenidas (y = Ai/Ap) en función de las relaciones de masas teóricas (x = mi/mp), donde Ai y mi son el área y masa del compuesto i y Ap y mp son el área y la masa del patrón interno. Además, se realizaron pruebas estadísticas de linealidad y proporcionalidad, debiendo cumplirse los criterios siguientes: coeficiente de correlación (r) de la ecuación de regresión ≥ 0,99; CV de los factores de respuesta, definidos como y/x (CVf) < 5 %, CV de la pendiente (CVb) < 2 % y los límites de confianza del intercepto debían incluir el cero (ICH, 2000; CECMED, 2014).
La Exactitud del método se determinó a partir del ensayo de linealidad mediante el recobrado a las concentraciones baja (50 %), media (100 %) y alta (150 %). Se aplicó la prueba estadística t de Student, para determinar si había diferencias significativas entre el recobrado promedio obtenido y el 100 %, según la cual, si la t exp < t tab no existen diferencias significativas entre el recobrado promedio obtenido y el 100 % (Castro et al., 1989).
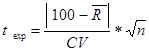
donde: 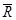 = recobrado promedio obtenido; n= número de réplicas y CV= coeficiente de variación (%)
= recobrado promedio obtenido; n= número de réplicas y CV= coeficiente de variación (%)
Además, se aplicó la prueba de G de Cochran para determinar si la concentración influye en la variabilidad de los resultados (ICH, 2000; CECMED, 2014), según la cual, si G exp < G tab la concentración no influye en la variabilidad de los resultados (Castro et al., 1989).
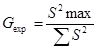
donde: S2max= desviación estándar máxima al cuadrado, ∑S2= desviación estándar total al cuadrado.
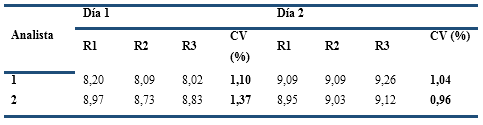
La Precisión se determinó mediante los ensayos de repetibilidad (un mismo analista analizó 10 réplicas de la misma muestra, en el mismo día y equipo) y de precisión intermedia (dos analistas, en dos días, analizaron tres réplicas independientes del mismo lote de crema). Se calcularon los CV (%) de la determinación del C28, C30 y del contenido total, y se halló, además, el límite de confianza (LC = Media ± t x DE) de la determinación del contenido total de alcoholes para p = 0,05. Para determinar si hubo diferencias significativas, se realizó una prueba ANOVA de dos vías (p = 0,05) (ICH, 2000; CECMED, 2014). Además, se consideró como criterio de aceptación que los CV de la Repetibilidad y de la Precisión intermedia fueran < 5,7 % (Castro et al., 1989).
La Especificidad del Método se determinó inicialmente por el análisis de muestras de cremas preparadas sin adición de patrón interno, de cremas placebo y de una muestra de 1-eicosanol (patrón interno), con vistas a determinar posibles interferencias. Además, se sometieron muestras a condiciones extremas, para determinar la especificidad del método ante la posible presencia de productos de degradación, según el procedimiento que se indica a continuación.
Hidrólisis ácida e hidrólisis básica: 1g de crema se colocó en tubo de ensayos, se le añadió 10 mL de ácido clorhídrico (o hidróxido de sodio) 0,1 mol/L y se selló. El tubo de ensayos se colocó en un envase seguro a 108 (C durante 24 horas y luego el contenido se trasvasó a un embudo separador, donde se le añadió agua para eliminar el reactivo utilizado. Se separó la fase acuosa de la orgánica, y la orgánica se preparó según lo descrito en la metodología analítica desarrollada.
Oxidación: 1g de crema se colocó en un tubo de ensayos, se le añadió 3 mL de peróxido de hidrógeno al 30 % y se selló. Se colocó en un envase seguro protegido de la luz, y se mantuvo a temperatura ambiente durante 7 días. Transcurrido este tiempo, la muestra se trasvasó a un embudo separador y se le añadió agua, para eliminar el reactivo. Posteriormente, se separó la fase orgánica de la acuosa y la orgánica se preparó según lo descrito en la metodología analítica desarrollada.
Fotólisis: 1g de crema se colocó en un tubo de ensayos, se selló y se colocó en una cabina con luz ultravioleta (254 nm) a temperatura ambiente durante 7 días, y se preparó según lo descrito en la metodología analítica desarrollada.
Termólisis: 1g de crema se colocó en un tubo de ensayos, se selló, se colocó en un envase seguro, a 108 (C durante 7 días, y se preparó según lo descrito en la metodología analítica desarrollada.
Todos los ensayos se realizaron por triplicado. La especificidad se comprobó por comparación de los cromatogramas correspondientes a las muestras sometidas a condiciones extremas con los de la muestra original sin degradar (ICH, 2000; CECMED, 2014). En ningún caso los tiempos de retención del patrón interno y de los excipientes debían coincidir con los del C28 y C30, cuyos picos deben poder ser integrados sin interferencias, aun en presencia de las señales cromatográficas que pudieran aparecer debido a procesos de degradación.
RESULTADOS Y DISCUSIÓN
Los resultados del ensayo de Aplicabilidad del Sistema (Tabla 1) permitieron conocer que la resolución promedio entre los picos cromatográficos del C28 y C30 fue mayor que el límite de aceptación (2,5); lo cual garantizó una separación adecuada entre estos compuestos. Además, la determinación del contenido de ambos alcoholes en seis réplicas de inyección presentó un CV menor que su límite de aceptación (0,5 %), todo lo cual permitió comprobar que el sistema instrumental se encontraba en condiciones para realizar la validación.
En el ensayo de Linealidad, la ecuación de regresión para el contenido total de alcoholes fue y = 0,8413 x + 0,0033, para el 1-octacosanol fue y = 0,9043 x - 0,0002 y para el 1-triacontanol fue y = 0,7856 x - 0,0001; con r = 1 en todos los casos, cumpliendo con el límite de aceptación (0,99). El CV de los factores de respuesta para el contenido total de alcoholes (CVf = 0,16 %) y para los alcoholes 1-octacosanol y 1-triacontanol (CVf = 0,03 %), cumplieron con el límite de aceptación establecido (5 %). El CVb fue de 0,001 % para el contenido total de alcoholes y de 0,0001 % para el C28 y C30, resultando inferiores que el límite de aceptación (2 %). El límite de confianza del intercepto, incluyó el cero en los tres casos, lo que indica que no existe sesgo. Teniendo en cuenta que estos resultados cumplen con los criterios exigidos, se puede afirmar que el método es lineal y proporcional en el intervalo de concentraciones estudiado.
La Exactitud del Método se determinó a partir del estudio de linealidad y los resultados se expresaron en forma de porcentaje de recuperación (Tabla 2). Al aplicar la prueba t de Student (ICH, 2000; CECMED, 2014), se demostró que no existen diferencias significativas entre la recuperación media y el 100 %, al ser la t exp (1,280) menor que la t tab (2,306) (p = 0,05, GL= 9-1), lo cual confirma que el método es exacto. Al aplicar la prueba G de Cochran se encontró que la G exp (0,616) es menor que la G tab (0,8709) (p = 0,05, n=3), por tanto, se puede concluir que, en el intervalo de concentraciones estudiado, la concentración no tiene influencia en la variabilidad de los resultados.
Tabla 2 Resultados del estudio de exactitud

*Con respecto a la concentración nominal de los alcoholes
Para determinar la Precisión del método se realizó inicialmente un ensayo de Repetibilidad, en el que la cuantificación de los alcoholes en la crema presentó un CV (1,41 %) inferior al establecido por Kolthoff (5-10 %) y por Horwitz (5,7 %), para las determinaciones cromatográficas de compuestos al 1 % (Castro et al., 1989), con límites de confianza entre 8,22 y 8,76 mg/g de crema. Estos resultados confirman que el método es repetible.
En el estudio de Precisión intermedia, el CV global de las 12 determinaciones (4,92 %), incluyendo los dos analistas (Tabla 3), fue inferior al establecido. A su vez, las determinaciones en los 2 días (n = 3) por los dos analistas, mostraron CV menores que 5,7 %, por tanto, el método es reproducible con límites de confianza entre 7,83 y 9,73 mg/g de crema, los cuales abarcan el intervalo de confianza determinado en el ensayo de repetibilidad. El análisis estadístico demostró que no existen diferencias significativas (p > 0,05) entre los resultados de cada analista y día, al utilizar la prueba ANOVA, lo cual indica que estos factores no influyen en la precisión del método. Teniendo en cuenta los resultados de la repetibilidad y la precisión intermedia, podemos afirmar que el método es preciso.
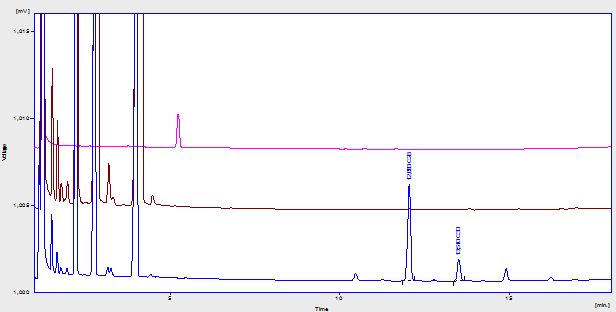
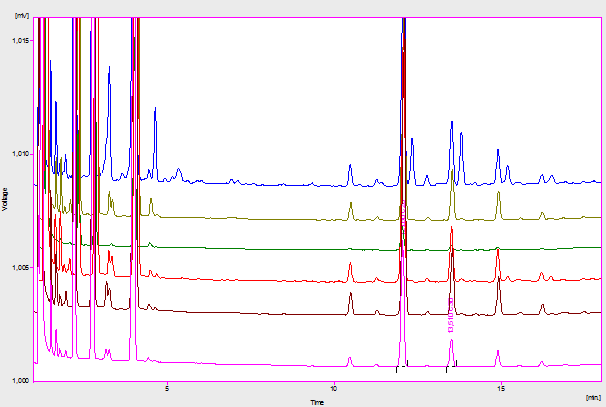
El estudio de la Especificidad permitió conocer que los alcoholes C28 y C30 pueden ser determinados sin interferencia del patrón interno (1-eicosanol) ni de otros compuestos presentes en la crema (Figura 1). Al analizar las muestras sometidas a condiciones de estrés, propicias a la ocurrencia de procesos de degradación (Figura 2), los picos extra que aparecieron no interfirieron con los analitos de interés, según se pudo comprobar por Cromatografía de Gases acoplada a Espectrometría de Masas, análisis realizado según Marrero et al., 2012. Lo anterior evidencia que el método es específico, incluso ante muestras potencialmente degradadas, por lo que puede ser empleado en estudios de estabilidad y en los controles de calidad.
CONCLUSIONES
El método analítico propuesto para determinar el contenido de 1-octacosanol y 1-triacontanol en la crema cumple con todos los requisitos establecidos, por lo que se considera validado. Se comprobó que es exacto, lineal y preciso en los intervalos de concentración estudiados, y también es específico aun para las muestras sometidas a condiciones de degradación. Por tanto, el método puede ser utilizado para el control de calidad y los estudios de estabilidad de la crema.

