










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión On-line ISSN 1561-2988
Rev Cubana Farm vol.49 no.2 Ciudad de la Habana abr.-jun. 2015
ARTÍCULO ORIGINAL
Validación de un método cromatográfico para la determinación de cafeína en muestras acuosas de la Industria Farmacéutica
Validation of a chromatographic method for determination of caffeine content in aqueous samples of the drug industry
Lic. Daylín Hernández Falcón, Dr. C. Oscar Ernesto Ledea Lozano, Dra. C. Lidia Asela Fernández García, Téc. Elena González García
Centro Nacional de Investigaciones Científicas (CENIC). La Habana, Cuba.
RESUMEN
Introducción: la cafeína es una sustancia que se encuentra en algunas plantas como el café, el té, el cacao, además es un ingrediente de algunos medicamentos como los analgésicos, es estimulante metabólico y del sistema nervioso central. Su consumo agudo o crónico puede dar lugar a una variedad de efectos adversos. Debido a su uso en la industria farmacéutica su determinación adquiere gran importancia. La validación de un método analítico demuestra científicamente que es adecuado para una aplicación específica, como la determinación de ingredientes farmacéuticos activos en disoluciones acuosas.
Objetivo: validar un método analítico para la cuantificación por Cromatografía Líquida de Alta Resolución de la cafeína presente en disoluciones acuosas y la determinación de su contenido en muestras residuales acuosas de un laboratorio farmacéutico productor de medicamentos.
Métodos: la validación del método cromatográfico se llevó a cabo empleando una columna RP-18 de 250 × 4,6 mm, 5 µm; Fase móvil: agua—CH3OH (70/30); flujo: 1,0 mL/min y un detector ultravioleta visible a 254 nm. La determinación de la cafeína se efectuó por el método validado según las regulaciones internacionales vigentes.
Resultados: el método resultó selectivo frente a los productos de ozonización, lineal (r= 0,999 y r2= 0,999), preciso, exacto, robusto y sensible, en el intervalo de 1 × 10-4 mol/L a 2 × 10-3 mol ̸ L. Se evaluó la estabilidad del analito en las condiciones de análisis. El contenido de cafeína en aguas residuales de la industria farmacéutica productora del medicamento fue determinado en concentraciones inferiores a 3 × 10-4 mol/L.
Conclusiones: el método validado fue selectivo, lineal, preciso, exacto y robusto en el intervalo de concentraciones analizado y permite la determinación de cafeína y del analito en disolución acuosa en muestras residuales acuosas del laboratorio farmacéutico productor del medicamento en Cuba.
Palabras clave: cafeína, cromatografía líquida de alta resolución, validación, determinación.
ABSTRACT
Introduction: caffeine is a substance located in some plants like coffee, tea, cocoa; it is also an ingredient of some drugs such as analgesics, a metabolic and central nervous system stimulant. The consumption of caffeine either acute or chronic may give rise to a variety of adverse effects. Due to its use in the drug industry, it is vitally important to determine it. The validation of an analytical method scientifically proves that it is adequate for a particular application as in the case of determination of active ingredients in aqueous solutions.
Objective: to validate an analytical method for the quantitation by means of high performance liquid chromatography of the caffeine content in aqueous solutions and in wastewater samples from a drug manufacturing laboratory.
Methods: the validation of the chromatographic method was performed by using a RP—18 de 250 x 4,6 mm, 5 µm column, a mobile phase: water/ CH3OH (70/30); Flow: 1,0 mL/min and an ultraviolet detector at 254 nm. The caffeine content was estimated by the high performance liquid chromatograpy following the international regulations in force.
Results: the method was selective against the ozonization products, linear r = 0.999 y r2 = 0.999), precise, accurate, robust and sensitive in the 1x10-4mol/L to 2x10-3mol/L interval. The analyte stability was also evaluated under the analysis conditions. The caffeine content in wastewaters of the drug manufacturing industry was determined at concentrations lower than 3x10-4 mol/L.
Conclusions: the validated method was selective, linear, precise, accurate and robust in the analyzed interval of concentrations and allows estimating the caffeine content in aqueous solutions and of the analyte in wastewater samples from a pharmaceutical laboratory that produces the drug in Cuba.
Keywords: caffeine, high performance liquid chromatography, validation, determination.
INTRODUCCIÓN
La cafeína (1,3,7‒trimetilxantina), también conocida como teína, mateína, guaranína, metilteobromina o metilteofilina, es un derivado del grupo de las xantinas y a su vez se deriva de las purinas. La fórmula química de la cafeína es C8H10N 4O2, con una masa molecular de 194,19 g/mol.1
La cafeína es una sustancia que se encuentra en ciertas plantas naturales como el café, el té, el cacao y en los chocolates y algunos refrescos. Por otro lado, la cafeína es un estimulante metabólico y del sistema nervioso central y un componente en cientos de medicamentos como analgésicos. Sus principales efectos son psicoestimulantes, cardiovasculares y broncodilatadores. El consumo agudo o crónico de cafeína puede dar lugar a una amplia variedad de efectos adversos e intoxicaciones. A pesar de que la cafeína no es carcinógena su producto de hidrólisis, la cafeidina, en humanos puede causar cáncer.1 Además, la cafeína disminuye la depuración de la teofilina e inhibe de forma competitiva el metabolismo de la clozapina pudiendo aumentar la probabilidad de aparición de efectos adversos.2 La cafeína puede considerarse un fármaco, un nutriente y una droga de abuso dependiendo de su uso.
La presencia de cafeína ha sido reportada en diferentes tipos de aguas. En aguas residuales domésticas se han detectado entre 20 y 300 µg ̸ L mientras que en los ríos se ha determinado 1500 ng ̸ L.3 La determinación de cafeína adquiere mucha importancia para el control de calidad de los productos que la contienen, debido a su uso en la industria de alimentos y en la industria farmacéutica.
Las técnicas analíticas más empleadas en la actualidad pueden englobarse en dos grandes grupos: técnicas de separación y técnicas espectroscópicas. Se han reportado varios métodos para la determinación de cafeína y sus productos, como los titrimétricos, espectofotométricos y la polarografía.4,5 Estos métodos tienen como desventaja la necesidad de grandes cantidades de muestras, procedimientos complicados o tediosos de preparación de muestras, además de no permitir la cuantificación de cafeína en presencia de otros compuestos. En la actualidad, la determinación de la cafeína se suele llevar a cabo mediante técnicas de separación como Cromatografía Líquida de alta Resolución (CLAR), Electroforesis Capilar, Cromatografía de Capa Fina y Cromatografía de Gases.6,7 Los métodos de CLAR usualmente emplean la determinación en fase reversa con detección mediante técnicas electroquímicas, ópticas y termoquímicas. Una desventaja que han presentado los métodos para la cafeína es que la misma muestra largos tiempos de retención, de 6 a 13 minuto.8,9 La elección de una u otra técnica depende del problema a resolver, del volumen de muestra y de su concentración.
La ejecución de un procedimiento de validación ofrece una demostración científica de que un método analítico es adecuado para una aplicación específica. Las agencias reguladoras han presentado varias guías para el procedimiento de validación como son: la Eurachem 1998,10 U.S. Food and Drug Administration (FDA; 2000),11 United States Pharmacopoeia (USP; 2009),12 International Conference on Harmonization (ICH; 2005)13 y la World Health Organization (WHO; 2006).14 Las guías oficiales no presentan una secuencia experimental para los procedimientos de validación debido a que la secuencia óptima puede depender del método específico a validar. Una secuencia para la validación de métodos analíticos cuantitativos de CLAR para el análisis de productos farmacéuticos es discutida por Bonfilio et al., 2012.15 El orden propuesto por los autores plantea el análisis de estabilidad de la disolución, selectividad, linealidad y rango, precisión (repetibilidad y precisión intermedia), exactitud, límite de detección, límite de cuantificación y robustez.
El objetivo de este trabajo es validar un método analítico de determinación de cafeína por CLAR y su empleo para la determinación del analito en aguas, debido al interés de estudiar la presencia de la cafeína en aguas residuales provenientes de plantas productoras de medicamentos en Cuba y para cuantificar la efectividad de la ozonización aplicada con el objetivo de eliminarla o disminuir su concentración en las aguas de vertimiento el tratamiento de las aguas residuales de este tipo de industria.
MÉTODOS
REACTIVOS
Los reactivos empleados para el desarrollo experimental fueron de grado analítico. Las disoluciones patrones de cafeína se prepararon a partir de un patrón de cafeína pura para análisis (99,5 %, Sigma, Código de Producto C0750). Se empleó Metanol (CH3OH) de la Merck para cromatografía líquida. Los ajustes de pH se realizaron con disoluciones de ácido fosfórico e hidróxido de sodio, ambos de la Merck y de calidad reactivo para análisis.
INSTRUMENTACIÓN
El análisis de la cafeína se llevó a cabo en un cromatógrafo líquido de alta resolución equipado con una bomba LKB 2150 (LKB, Bromma, Sweden), un inyector manual con loop de 20 µL (Rheodyne 7725i), un detector espectrofotométrico de longitud de onda variable Knauer K-2501 (Knauer, Germany), una interfase Knauer, que permite la adquisición de los cromatogramas mediante el software Eurochrom 2000 (Knauer, Germany). La determinación se realizó empleando una columna de fase reversa y en modo isocrático. Se empleó una mezcla agua ̶ metanol (70: 30 v ̸ v) como fase móvil y una columna RP 18 de 250 × 4 mm (5 µm), Grace Vydac. Se trabajó con una velocidad de flujo de 1 mL ̸ min. La longitud de onda para la detección UV (254 nm) se seleccionó tomando uno de los máximos de absorbancia en un espectro UV (200-600 nm) de una disolución de cafeína. Se empleó un espectofotómetro Cintral 101 para este fin.
El pH fue medido con un pHmetro-conductímetro InoLab. En todos los casos se realizaron las determinaciones a temperatura ambiente (25 °C).
Se emplearon dos programas de computación: Microsoft® Excel 2007 y el programa estadístico STATGRAPHICS Plus 5.0 para el procesamiento estadístico de los resultados.
PARTE EXPERIMENTAL
La validación de cafeína por CLAR, como un primer paso para la determinación de dicho analito en aguas, se lleva a cabo según la guía ICH de 2005, se tiene en cuenta el procedimiento descrito por Bonfilio et al., 2012;15 el cual es más específico para el desarrollo de métodos analíticos cuantitativos de CLAR. El método validado no se ha reportado previamente.
1. Estabilidad de la disolución: se estudió el tiempo y la temperatura en la cual la disolución de cafeína permanece estable para su posterior uso, mediante la medición de disoluciones patrones de 1 × 10-3 mol/L de cafeína a los 0, 1, 3, 5, 7, 14 y 21 días después de su preparación. Durante el estudio, se mantiene la disolución a temperatura ambiente y en refrigeración a 10 °C. Se realizó la prueba Anova para comparar las muestras y se obtuvo el gráfico de las medias por el procedimiento de diferencia mínima significativa (LDS) de Fisher para las dos condiciones de temperatura en los diferentes tiempos.
2. Selectividad: se preparó una disolución de cafeína de 1 × 10-3 mol/L y la misma fue sometida a procesos de oxidación mediante la ozonización a pH 7, se determina así la posibilidad de la aparición de interferencias en la determinación de la cafeína por la presencia de los productos de degradación. Además, se calculó la resolución (Rs) entre la cafeína y su producto más cercano.
Criterio de aceptación: no deben aparecer señales interferentes atribuidas a los posibles productos de degradación, que coincidan con el tiempo de retención del patrón del analito, o que estén muy cercanas a la zona de interés analítico para la cafeína. La Rs debe ser mayor que 2,5.
3. Linealidad y rango: los experimentos para la determinación de la linealidad se realizaron mediante el análisis de disoluciones estándar de cafeína por triplicado (0,1; 0,5; 0,8; 1; 1,2; 1,5; 2 × 10-3 mol/L) en un intervalo de 10-200 % de la cantidad teórica declarada como 100 % (1 × 10-3 mol/L). Se empleó el método de regresión simple para la obtención de la curva de calibración. Además, se determinó r (coeficiente de correlación lineal), r2 (coeficiente de determinación), a (intercepto) y b (pendiente), para el 99 % de confianza.
CRITERIOS dE aCEPTACIÓN
- Ecuación de la recta:
y= bx + a
Donde r ³ 0,99 y r2 ³ 0,98
- Prueba de hipótesis nula de la ordenada en el origen a= 0 y de la pendiente b= 0. Se determinó a partir de una prueba ANOVA de la regresión, teniendo en cuenta la probabilidad asociada al valor de la ordenada y la pendiente, es decir, si la p ˂ 0,05; el valor difiere significativamente de cero.
Además, la desviación estándar relativa de la pendiente (Sbrelativa) debe ser menor del 2 %.
Adicionalmente, se determinó el error absoluto medio (MAE) que es el valor medio de los residuos y el estadístico Durbin-Watson (DW) que examina los residuos.
Se estableció el intervalo en que se cumplieron los criterios de linealidad, exactitud y precisión del método estudiado.
4. Precisión (repetibilidad y precisión intermedia)
• Repetibilidad: se evaluaron muestras con la concentración equivalente al 100 %, un valor bajo y otro alto, comprendidos dentro del intervalo de la linealidad del método, 50 y 150 %; respectivamente. Se calculó el CV en estos tres niveles de concentración y se comparó con el criterio establecido. Las determinaciones fueron realizadas por sextuplicado por el mismo analista, en las mismas condiciones de trabajo. Criterio de aceptación: CV ≤ 1,5 %.
• Precisión intermedia: se analizaron muestras de 1 × 10-3 mol/L de cafeína, por sextuplicado, bajo las mismas condiciones y en el mismo laboratorio pero días diferentes por dos analistas diferentes. Se calculó el CV total. Criterio de aceptación: CV ≤ 2,0 %. Además, se realizó la prueba F de Fisher y la t de Student, para comparar varianzas y medias, respectivamente.
5. Exactitud: la exactitud fue determinada por el análisis por triplicado de muestras con cantidades conocidas de cafeína (0,5; 1 y 1,5 × 10-3 mol/L). La exactitud como porcentaje del recobrado fue evaluada, incluyendo la determinación del recobrado medio y del Coeficiente de Variación (CV) total.
Recobrado (R)= Concentración recobrado real ̸ Concentración añadida × 100
Criterios de aceptación: 97 % ≤ R ≤ 103 % y CV ≤ 2,0 %.
Además, se realizó la prueba G de Cochran y la prueba de la t de Student, con n-1 los grados de libertad y α= 0,05.
6 y 7. Límite de detección y Límite de cuantificación: el límite de detección (LD) y el límite de cuantificación (LC) se obtuvieron a partir de la curva de regresión lineal como 3,3 ∂/ (pendiente de la curva) y 10 ∂/ (pendiente de la curva), respectivamente, donde ∂ es la desviación estándar del intercepto. Ambos límites fueron comprobados experimentalmente mediante la medición de disoluciones de cafeína de concentraciones conocidas y cercanas a los límites calculados.
8. Robustez: se analizaron muestras de concentración 1 × 10-3 mol/L por triplicado y se evaluó la influencia de la variación de cuatro factores que pudieran incidir en los resultados, variando un solo parámetro a la vez, como son: variación del pH de la fase móvil en ± 0,2 unidades, variación de la composición orgánica de la fase móvil en ± 2 %, el empleo de otra columna de separación, Lichrospher® 100 RP18 (5 µm), Merck y la preparación de la muestras con otro tipo de agua, agua potable. Se realizó la prueba estadística de Fisher.
Criterio de aceptación: la probabilidad estadística de la prueba de Fisher debe ser superior a 0,05.
La determinación de cafeína fue llevada a cabo, con el método previamente validado, a muestras de aguas residuales obtenidas de una industria farmacéutica productora de ampolletas de Cafeína y Benzoato de Sodio en Cuba.
Muestreo y ozonización de las aguas residuales
Las muestras de las aguas residuales de la producción de cafeína en la industria fueron colectadas en el punto de salida de las aguas residuales, en tres oportunidades, durante la producción en campaña del medicamento. La conservación de las muestras hasta el momento de análisis se realizó a 10 °C en refrigeración. Se empleó un filtro (0,45 µm) para filtrar las muestras antes de ser inyectadas en el cromatógrafo.
La instalación empleada para el proceso de ozonización consistió en un reactor de vidrio de 100 mL de volumen, funcionando en semibatch, equipado con un difusor poroso de borosilicato, puertos para la extracción de la muestra, entrada y salida de gas. El reactor fue acoplado a un baño termostatado (Frigomix U-2, Alemania) que controla la temperatura a 25 ± 0,1 °C. La mezcla oxígeno—ozono se obtuvo con el empleo de un ozonizador (modelo AQOZO, CIOZONO, Cuba), a partir de oxígeno puro. El ozono en exceso a la salida del reactor fue destruido catalíticamente. La concentración de entrada de ozono gaseoso fue 45 mg/L, determinada a 256 nm espectrofotómetricamente (Ultrospec III, Pharmacia, UK). La ozonización de las muestras se realizó a pH 7.
RESULTADOS
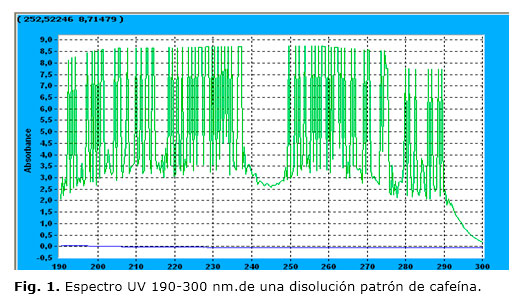
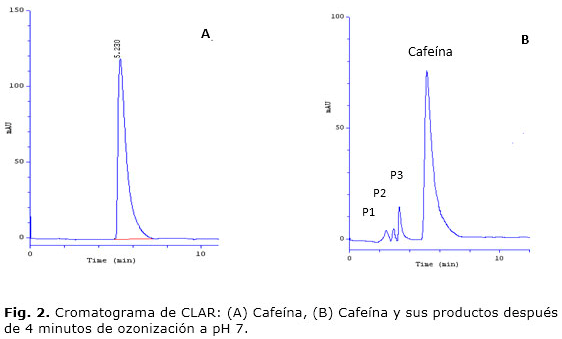
En la figura 1 se muestra el espectro UV obtenido para una muestra patrón de cafeína de concentración de 1 × 10-3 mol/L. En el mismo, se pueden observar varios máximos de absorción en el intervalo de 190 a 300 nm, mientras que la absorbancia en el intervalo de los 300-600 nm fue muy similar a la línea base. La longitud de onda seleccionada para los posteriores análisis por CLAR fue de 254 nm, con el objetivo de evitar posibles interferencias de otros compuestos carbonados que son sensibles a las menores longitudes de onda y de seleccionar uno de los máximos de absorbancia de mayor intensidad. En la figura 2 A se muestras un cromatograma típico de la cafeína obtenido con las condiciones estudiadas.
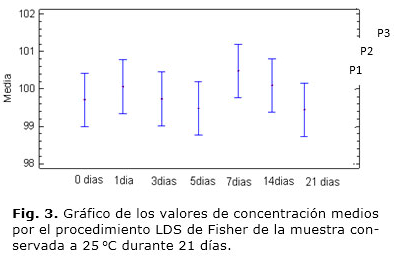
1. Estabilidad de la disolución: las muestras analizadas, a 10 y 25 °C, no dieron pruebas visuales ni en cuanto a las áreas de los picos, de degradibilidad o hidrólisis durante las primeras tres semanas después de preparadas y conservadas. El valor F de la tabla Anova fue de 0,96; con p= 0,44.
La figura 3 presenta el gráfico de las medias obtenido por LDS en el estudio realizado a 25 °C. Sin embargo, el CV del tiempo de retención de la cafeína mostró un incremento hasta un 5 % a partir de la segunda semana de realizado el análisis, fundamentalmente a 25 °C.
2. Selectividad: el cromatograma obtenido luego de la ozonización a pH 7 se muestra en la figura 2B, observándose el pico de la cafeína a los 5,23 minutos y tres productos de degradación P1, P2 y P3, con tiempos de retención entre los dos y cuatro minutos. La Rs calculada fue de 3,3.
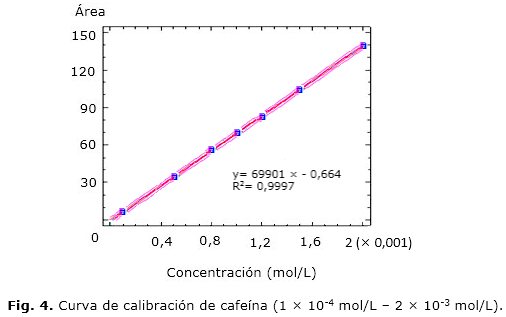
3. Linealidad y rango: la curva de calibración obtenida al graficar el área integrada bajo el pico cromatográfico contra la concentración de las muestras se presenta en la figura 4. Los valores estadísticos calculados se presentan en la tabla 1. El intervalo evaluado fue de 10-200 %.
4. Precisión, 5. Exactitud, 8. Robustez: los resultados correspondientes a la validación de este método para estos parámetros se resumen en la tabla 1, donde Fexp, texp, Ftab y ttab se refieren al valor respectivos de F Fisher y t de Student tabulados y determinados experimentalmente.
6. Límite de detección, 7. Límite de cuantificación: El límite de detección calculado fue 2,33 × 10-5 mol/L y el límite de cuantificación fue de 7,07 × 10-5 mol/L. Ambos límites se comprobaron experimentalmente con resultados satisfactorios.
La concentración de cafeína en las aguas residuales y ozonizadas fue determinada con el método propuesto. Los resultados obtenidos se presentan en la tabla 2.

DISCUSIÓN
El empleo de la secuencia de validación propuesta por Bonfilio y colaboradores, 201215 permitió la validación del método propuesto de una manera rápida y eficiente. Dicha secuencia incluye todos los parámetros regulados por la ICH, 200514 y aporta como ventaja una descripción más específica y organizada para el desarrollo de métodos por CLAR.
Estabilidad de la disolución: el valor p de la razón F de la Anova para la comparación entre las áreas de los picos cromatográficos es mayor que 0,05, por lo que no existe una diferencia estadísticamente significativa entre las medias de las variables con un nivel del 95,0 % de confianza. A pesar de estos resultados se recomienda el empleo de las disoluciones de cafeína en los primeros 15 días después de preparada, considerando las variaciones experimentadas en el tr del compuesto, que puede estar asociada a la degradación o hidrólisis del analito en disolución.
Selectividad: el método cromatográfico aplicado permite una adecuada separación del analito respecto a las posibles interferencias en el proceso de ozonización, a pesar de que los productos no fueron completamente separados entre sí. La Rs calculada corrobora este hecho. Por tanto, el método puede emplearse para la determinación de cafeína frente a sus productos de degradación P1, P2 y P3.
Linealidad y rango: el sistema mostró un curso lineal en el intervalo de 10-200 %, se cumplieron todos los criterios estadísticos establecidos al procesar los resultados por regresión lineal. Dado que la probabilidad estadística en la tabla Anova fue inferior a 0,01, la relación entre Área y Concentración fue estadísticamente significativa para un nivel de confianza del 99 %, mientras que la probabilidad estadística de la ordenada fue superior a 0,05; por lo que el valor de "a" no fue significativamente diferente de cero.
El coeficiente de determinación r2 indicó que el modelo explicó un 99,96 % de la variabilidad en Área. El coeficiente de correlación indicó una relación relativamente fuerte entre las variables. El error estándar de la estimación mostró que la desviación típica de los residuos fue 1,13. El valor medio de los residuos (MAE) fue de 0,77. Dado que la probabilidad estadística de DW fue superior a 0,05, no hubo indicio de una posible correlación serial.
El intervalo analítico en el que se cumplieron los criterios de aceptación de la validación fue de 10-200 %, verificándose que el método fue adecuado para su aplicación a muestras que contienen principio activo a concentraciones dentro del intervalo de concentración definido.
Precisión: la repetibilidad de la respuesta analítica del método fue demostrada mediante la obtención de CV bajos, inferiores en todos los casos al 1,5 % establecido como límite. El estudio de precisión intermedia también mostró un CV inferior al criterio de aceptación. Este análisis se complementó con las pruebas estadísticas de Fisher y la t de Student donde la Fexp ˂ Ftab y Texp ˂ Ttab, por lo que no existieron diferencias significativas entre las precisiones de los analistas, independientemente del día en que se efectúo el ensayo. El conjunto de dichos resultados permitió asegurar que estos fueron homogéneos y que los errores aleatorios no repercutieron apreciablemente, lo que ratifica la precisión del método.
Exactitud: el recobrado medio no excedió el límite de 97-103 % por lo que el método fue exacto. Además el CV total quedó comprendido en el intervalo de aceptación. La Gexp fue menor que Gtab de la prueba de Cochran, por lo que las varianzas de los tres niveles de concentración evaluados fueron equivalentes y no influyó el factor de concentración en la exactitud del método. Por su parte, la prueba de la t de Student corroboró la exactitud, ya que el valor obtenido fue inferior al tabulado, por lo que no existieron diferencias estadísticamente significativas entre el recobrado medio y el 100 %. Se puede afirmar que la técnica fue exacta y no se afectó por errores sistemáticos de forma significativa.
Límite de detección, Límite de cuantificación: el método fue suficientemente sensible para el propósito con que fue desarrollado, pues es capaz de detectar una cantidad equivalente al 2,30 % de la cantidad teórica declarada como 100 % frente a la degradación por ozonización del principio activo.
Robustez: la razón F de la prueba de Fisher para la comparación entre varias muestras, que en este caso es igual a 1,64, es el cociente entre el desviación entre grupos y la desviación dentro de grupos. Puesto que la probabilidad estadística p es mayor que 0,05, no existe una diferencia estadísticamente significativa entre las medias de las variables con un nivel del 95,0 % de confianza, demostrando la robustez del método respecto a los cuatro parámetros variados.
ANÁLISIS DE MUESTRAS DE AGUAS RESIDUALES DE LA INDUSTRIA FARMACÉUTICA
La presencia de cafeína en las aguas residuales provenientes de la industria farmacéutica fue confirmada, mediante la detección y cuantificación del analito en las muestras colectadas por el método validado. El contenido presente en las muestras estuvo dentro del intervalo definido para hacer la validación del método en todos los casos. El resultado obtenido era de esperarse como consecuencia de los procesos de producción y limpieza en la industria que generan gran cantidad de aguas residuales contaminadas con cafeína. Las muestras tratadas mediante la ozonización a pH 7 presentaron cantidades inferiores al límite de detección, debido a la degradación de la cafeína frente al tratamiento con ozono.
El método validado fue selectivo, lineal, preciso, exacto y robusto en el intervalo de concentraciones que se analizan y permite la determinación de cafeína en disolución acuosa y en muestras residuales acuosas del laboratorio farmacéutico productor del medicamento en Cuba.
REFERENCIAS BIBLIOGRÁFICAS
1. International Food Information Council Foundation. Caffeine & Health: Clarifying the controversies. IFIC Review 2008. [Citado: 4 de enero de 2014]. Disponible en: http://www.ific.org
2. Kari R, Virpi R, Jouko L, Pertti N. Effect of Caffeine- Containing versus Decaffeinated Coffee on Serum Clozapine Concentrations in Hospitalised Patients Basic. Clin Pharmacol Toxicol. 2004;94:13-8.
3. Brown GK, Zaugg SD, Barber LB. Wastewater analysis by gas chromatography / mass spectrometry. Water Resources Investigations. Rep. U.S. Geol Surv. 1999;99 4018B:431-5.
4. Wanyika HN, Gatebe EG, Gitu LM, Ngumba EK, Maritim CW. Determination of caffeine content of tea and instant coffee brands found in the Kenyan market. African Journal of Food Science. 2010;4(6):353-358.
5. Komes D, Horžić D, Belščak A, Kova K and Baljak A. Determination of Caffeine Content in Tea and Maté Tea by using Different Methods. Czech J. Food Sci. Special Issue. 2009;27.
6. Švorc Ľ. Determination of Caffeine: A Comprehensive Review on Electrochemical Methods. Int. J. Electrochem. Sci., 2013;8:5755-73.
7. Weimann A, Sabroe M, Poulsen HE. Measurement of caffeine and five of the major metabolites in urine by high-performance liquid chromatography/tandem mass spectrometry. J. Mass Spectrom. 2005;40:307-316.
8. Patil, Pandurang N. Caffeine in various samples and their analysis with HPLC - A review. International Journal of Pharmaceutical Sciences Review & Resear. 2012;16(2):76.
9. Anand patil, varun raheja, anagha cafeínamre. Simultaneous analysis of intestinal permeability markers, caffeine, paracetamol and sulfasalazine by reverse phase liquid chromatography: a tool for the stancafeínardization of rat everted gut sac model. Asian Journal of Pharmaceutical and Clinical Research. 2010;3(3):204-7.
10. EURACHEM Guide: The Fitness for Purpose of Analytical Methods. A Laboratory Guide to Method Validation and Related Topics. First English Edition 1.0; 1998.
11. U.S. Food and Drug Administration. FDA Technical Review Guide; Center for Drug Evaluation and Research (CDER), Guidance for Industry, Analytical Procedures and Methods Validation, Chemistry Manufacturing and Controls Documentation, Draft Guidance; 2000. [Citado: 4 de enero de 2014]. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidance/ucm122858.pdf
12. The United States Pharmacopoeia. United States Pharmacopoeial Convention: Rockville, Md; 2009.
13. ICH –Harmonised tripartite Guideline. Validation of analytical Procedures: Text and Methodology Q2 (R1); 2005.
14. WHO- Technical Report Series No. 937. Appendix 4n Analytical Method Validation; 2006.
15. Bonfilio R, Laignier EC, Benjamim M, Nunes HR. Analytical Validation of Quantitative High-Performance Liquid Chromatographic Methods in Pharmaceutical. Analysis: A Practical Approach, Critical Reviews in Analytical Chemistry. 2012;42(1):87-100.
Recibido: 11 de febrero de 2014.
Aprobado: 3 de septiembre de 2014.
Daylín Hernández Falcón. Centro Nacional de Investigaciones Científicas (CENIC). Ave. 25 esq. 158, Cubanacán, Playa, La Habana, Cuba. Teléfono: 208-3654.


{kind=link}