










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Medicina
versión On-line ISSN 1561-302X
Rev cubana med v.46 n.1 Ciudad de la Habana ene.-mar. 2007

Hospital Clinicoquirúrgico "Hermanos Ameijeiras"
Hígado graso. Enfoque diagnóstico y terapéutico
Dr. Alfredo Herrera González,1 Dr. Alfredo Nasiff Hadad,2 Dr. Enrique Arus Soler,3 Dr. Cosme Cand Huerta2 y Dra. Nancy León4
Resumen
El enfoque diagnóstico y terapéutico que se propusieron en esta revisión sobre "hígado graso" se hizo en función de las necesidades del médico práctico. Se planteó una disquisición teórica sobre el problema terminológico de este síndrome y se abordaron los mecanismos patogénicos comunes en las diferentes condiciones clínicas que predisponen a padecer este problema de salud que puede presentarse como una simple esteatosis hepática, pero también puede llegar a ser causa de una cirrosis hepática que conduzca al paciente a un transplante hepático. Se consideró la importancia de sopesar la suma de factores que predisponen a la enfermedad, los estudios imagenológicos y marcadores de riesgo de desarrollo de fibrosis antes de indicar la biopsia hepática. El tratamiento va dirigido a disminuir el flujo de ácidos grasos al hígado, proteger al hepatocito de mecanismos oxidativos, evitar tóxicos conocidos que dañen al hígado y tratar los factores condicionantes o asociados. Se propuso un algoritmo de atención y seguimiento de estos enfermos.
Palabras clave: Enfermedad grasa no alcohólica del hígado, esteatosis simple, esteatohepatitis, enfermedad metabólica.
La aparición del ultrasonido diagnóstico ha provocado que algunas enfermedades sean detectadas cuando aún se encuentran asintomáticas, de tal manera que estos hallazgos imagenológicos constituyen el motivo de la concurrencia del paciente a la consulta médica.
Una de estas situaciones médicas es la entidad identificada por los pacientes como grasa en el hígado o hígado graso, conocida más, hasta hace algunos años, por la teoría que por la necesidad de su abordaje en la práctica clínica.
Se estima que entre 15 y 39 % de los pacientes sometidos a biopsia hepática padecen esta enfermedad.1 En series de autopsias de pacientes fallecidos a causa de accidentes, la prevalencia de hígado graso es de 16 a 24 %.2 En población general de Italia y Japón se observó en la ecografía hígado graso en 16,4 y 23 %, respectivamente.3 Las estimaciones actuales de prevalencia de hígado graso en la población general en Estados Unidos son de 25 %.4
Se han planteado muy diversas interpretaciones acerca de las causas, la evolución, el pronóstico, las orientaciones dietéticas, el uso o no de medicamentos y la efectividad de las drogas; por lo que nos preguntamos: ¿Cuáles serían los mecanismos patogénicos de la enfermedad de acuerdo con las condiciones clínicas asociadas a ella? ¿Cuáles serían los pasos a seguir para el diagnóstico confirmatorio de hígado graso? ¿El grado de severidad de la enfermedad determinaría su evolución? ¿Se puede, mediante la terapéutica, modificar la historia natural de esta enfermedad?
Para dar respuesta a estas interrogantes fue convocado un grupo multidisciplinario del Hospital Clinicoquirúrgico Hermanos Ameijeiras para poner al día la información existente acerca de la etiología, diagnóstico y tratamiento del hígado graso. El presente artículo resume este análisis.
El problema terminológico
La terminología científica debe cumplir ciertos requisitos lingüísticos para servir a su finalidad fundamental de colaborar al desarrollo. La ambigüedad es, sin duda, uno de sus defectos esenciales junto a una significación polisémica de los términos. Estos últimos problemas los enfrenta la terminología relacionada con el tema que nos ocupa. La variedad de términos usados para describir esta entidad no deja claro a qué nos estamos refiriendo. Hay términos que se usan como sinónimos a pesar de sus diferentes significados clínicos, pero la sanción del uso los trasmite de generación a generación sin que haya una preocupación por buscar un término adecuado. El síndrome clínico-patológico que presentamos se conoce por hepatitis del hígado graso, hepatitis de la diabetes, esteatohepatitis no alcohólica o esteatosis hepática. El primero en utilizar el término "esteatohepatitis no alcohólica" (EHNA) fue Ludwig,5 en 1980, para describir los peculiares hallazgos histológicos en la hepatitis no alcohólica en pacientes que no tenían este hábito tóxico.
Los cambios histopatológicos que se observan en el síndrome clínico-patológico conocido como hígado graso (del inglés, fatty liver), son similares en disímiles condiciones clínicas que llevan al médico a interpretar su existencia y hacen necesario el diagnóstico etiológico (alcoholismo, drogas, pacientes no alcohólicos con factores predisponentes como ser obeso por ejemplo, diabéticos tipo 2, hiperinsulinémicos, dislipidemias tipo IV, malnutrición, o de causa desconocida).6 Cuando excluimos la etiología alcohólica y las otras causas de enfermedad hepática crónica (enfermedades hepáticas autoinmunes, enfermedad de Wilson, hepatitis C), nos quedamos con los cambios clínico-patológicos descritos, en inglés, como non-alcoholic fatty liver disease (NAFLD), término sustituido arbitrariamente por esteatosis o esteatohepatitis no alcohólica (EHNA), como sinónimos. Estos 2 últimos términos son en realidad parte de un espectro de formas histológicas de la enfermedad (NAFLD), que nosotros la denominaremos, en lo siguiente, enfermedad grasa no alcohólica del hígado (EGNAH). La finalidad de esta precisión es evitar la confusión de esta última con sus formas de presentación histopatológicas.
Patogenia
Hipótesis que tratan de explicar la patogenia de la EGNAH:
- Teoría de los 2 golpes.7 Explica los cambios histológicos ocurridos por 2 golpes que expresan 2 mecanismos sucesivos, "primera agresión" es la acumulación grasa en el hepatocito8 y "segunda agresión" se relacionaría con el estrés oxidativo, la peroxidación de los lípidos, la producción de aldehído malónico, de 4-hidroxinonenal, de citoquinas proinflamatorias, la activación de la células estrelladas del hígado y el estímulo de la fibrogénesis.9
La acumulación grasa ocurre por varias condiciones que favorecen este mecanismo:
- Aumento del flujo de ácidos grasos al hígado, que favorece su captación. La fuente de estos triglicéridos es endógena (tejido adiposo) y exógena (absorción intestinal).10
- Incremento de la síntesis endógena de triglicéridos por la esterificación de los ácidos grasos captados por el hepatocito.11
- Disminución de la secreción de las lipoproteínas de muy baja densidad (VLDL).12
- Disminución de la betaoxidación mitocondrial de ácidos grasos.13-15
La lipoperoxidación ocurre por aumento de la producción de radicales libres que favorecen el estrés oxidativo. Este último proceso es seguido por la activación de la cascada inflamatoria y de células que intervienen en la inflamación.16,17
- Variabilidad genética.18 Dada la variabilidad biológica y las diferencias del desarrollo progresivo de la enfermedad, se ha planteado el mecanismo de la variabilidad genética donde se demuestran diferentes polimorfismos genéticos (polimorfismo genético del citocromo microsomal P450 2E1, 4A47, 4A48,19 del promotor del factor de necrosis tumoral alfa (TNF-a), o del promotor de la interleuquina 10 y los polimorfismos de genes que codifican las proteínas implicadas en la generación de metabolitos reactivos de oxígeno,20 las defensas antioxidantes y las citocinas).
- Sobrecrecimiento bacteriano en el intestino delgado.21 Es la hipótesis que plantea un sobre crecimiento bacteriano en el intestino delgado, lo cual provoca una endotoxemia, llegando las toxinas al hígado por vía portal y estas desencadenan elevadas concentraciones del TNF-a. Si a esto le sumamos una aumento de la sensibilidad a endotoxinas y citoquinas,22 es mayor el daño directo de ellas en el hepatocito lo cual eleva la producción de radicales libres. Todo lo anterior también favorece el estrés oxidativo.
- La insulinorresistencia.23 La EGNAH se asocia estrechamente con el síndrome de resistencia a la insulina. Este fenómeno favorece la acumulación de ácidos grasos libres en el hígado. Ellos dentro del hepatocito son sustratos e inductores de las lipoxigenasas del citocromo mitocondrial, aumentan los niveles de citocromo P450 2E1, por consiguiente se eleva la producción de radicales libres. Igualmente, se predispone de esta manera al estrés oxidativo y se favorece la peroxidación de los lípidos de membrana del hepatocito. Se conoce además que el TNF-a desempeña un papel importante en los mecanismos moleculares de la resistencia a la insulina para el desarrollo de la EGNAH. La obesidad central se ha asociado con altos niveles de TNF-a, la misma se refleja por un mayor índice cintura/cadera (considerado como predictor de esteatosis hepática).24
Estos son los mecanismos básicos, comunes a las diferentes condiciones predisponentes para la génesis de la EGNAH. La combinación de varios mecanismos patogénicos se pueden evidenciar en los modelos siguientes:
En la diabetes tipo 2, aumentan los ácidos grasos libres captados por el hepatocito que se asocian al mecanismo de la insulinorresistencia. Así mismo se incrementa el CYP2 E1 y puede existir, al mismo tiempo, un síndrome de sobrecrecimiento bacteriano que provoca un elevado nivel de TNF-a.
En la obesidad, se suman, al aumento de los ácidos grasos libres, dependiendo de la insulinorresistencia, la susceptibilidad al daño hepático producido por endotoxinas y por un incremento del TNF-a . En el caso del síndrome de sobrecrecimiento bacteriano causado por bypass yeyunoileal o gastroplastia que son cirugías indicadas como tratamiento de la obesidad, se genera el paso de toxinas, citoquinas al sistema porta y se desencadenan así los efectos del TNF-a sobre el hepatocito. Cuando por el contrario se produce una pérdida brusca de peso, se predispone al desarrollo de EGNAH por un aumento de la lipólisis que trae aparejado un incremento de los ácidos grasos libres, y de los niveles de CYP2 E1, además, de la depleción del glutatión mitocondrial.
Histopatología
La esteatosis hepática puede ser microvacuolar o macrovacuolar,25 en dependencia del tamaño de las vacuolas grasas. Los triglicéridos son su componente principal, aunque pueden acumularse varias clases de lípidos.
La esteatosis microvacuolar consiste en la aparición de pequeñas vacuolas grasas citoplasmáticas. Las células pueden o no estar agrandadas. Ocurre en una variedad de enfermedades genéticas y adquiridas, incluyendo enfermedades metabólicas, el hígado graso del embarazo, el síndrome de Reye, la degeneración espumosa del alcoholismo y reacciones hepatotóxicas a drogas y toxinas.
En la esteatosis macrovacuolar, los hepatocitos comprometidos son de mayor tamaño que el resto. La fusión de vacuolas produce quistes grasos, a veces acompañados de mínima inflamación, en forma de lipogranulomas. Es la lesión clásica vinculada al alcoholismo crónico y se encuentra en gran número de condiciones, como la desnutrición/ kwashiorkor , la nutrición parenteral, la obesidad, la diabetes tipo 2, la hepatitis C, el bypass yeunoileal, etc.
En muchos casos, generalmente en los más severos, existe un patrón mixto de esteatosis, macrovacuolar y microvacuolar. Existen múltiples clasificaciones que intentan describir los diferentes grados y estadios. Expondremos una de ellas, la cual consideramos más útil para el médico en la práctica clínica.
La infiltración grasa del hígado tiene diferentes grados que van desde la esteatosis simple, pasan por la esteatohapatitis hasta la fibrosis y/o cirrosis. Cada uno de ellos puede tener un estadio según su severidad (porcentaje de hepatocitos afectos) en leve, moderada y severa.26
La distinción entre la etiología alcohólica y la no alcohólica desde la histopatológica lo establecen más criterios cuantitativos que cualitativos ya que en la forma alcohólica son más abundante los cuerpos hialinos de Mallory, hay mayor densidad del infiltrado polimorfonuclear y grados avanzados de fibrosis, por ello se recomienda que el diagnóstico histopatológico sea el de esteatohepatitis y que su causa sea determinada por los antecedentes de alcoholismo del paciente.
Diagnóstico. Ruptura de los estereotipos clínicos
La mayoría de los pacientes con EGNAH no complicada están asintomáticos, pero pueden sufrir molestias abdominales vagas, predominantemente en el hipocondrio derecho, y decaimiento. Muchos de ellos están entre los 40 y 60 años de edad aunque también se reporta en personas jóvenes.27 En raras ocasiones, aparece ictericia de tipo colestásico intrahepático.28 El examen físico es generalmente normal, con excepción de una hepatomegalia palpable blanda de borde romo.29
En todo paciente con sospecha de EGNAH debemos hacer un estudio de la función hepática. Las alteraciones bioquímicas consisten en una elevación de leve a moderada de las transaminasas, en particular la alaninoaminotransferasa (ALAT).30 En muchos casos, se plantea que la ALAT puede subir de 2 a 5 veces su valor normal. Por tanto, existe una relación aspartatoaminotransferasa (ASAT)/ALAT menor que 1.31 Cuando el índice ASAT/ALAT32 es inferior a 2 y mayor que 1, sugiere mayor probabilidad para el desarrollo de una forma progresiva de la enfermedad. En cambio, la existencia de una relación ASAT/ALAT mayor que 2 permite orientar el diagnóstico a una etiología alcohólica. Otras enzimas utilizadas como marcadores son la γ-glutariltransferasa33 (se eleva 2 veces el valor normal) y la fosfatasa alcalina. Se deben realizar marcadores virales para la hepatitis B y C para descartar portadores asintomáticos y determinación de triglicéridos ya que la hipertrigliceridemia constituye un factor de riesgo de EGNAH.
El hígado graso puede detectarse también con técnicas imageneológicas,34 como ecografía, tomografía axial computadorizada y resonancia magnética nuclear; cada una de estas técnicas con diferentes porcentajes de sensibilidad y especificidad. La ecografía constituye en nuestro medio el examen que con más frecuencia descubre un hígado graso asintomático.
El perfil clínico "típico" es el de una mujer de edad mediana, obesa, con diabetes tipo 2 o sin ella, dislipidemia e hipertensión.35 En ocasiones, se ha asociado también con algunas drogas, como estrógenos sintéticos a altas dosis, metotrexate, amiodarona y tamoxifeno. La obesidad es, sin duda, la condición clínica más frecuente y se describe en 86 % de los casos.36 Otros estudios han demostrado que el perfil de pacientes puede ser mucho más amplio que el inicialmente planteado. Bacon y otros,37 en una serie de pacientes con EGNAH reportaron que 58 % de los pacientes eran varones, 61 % no eran obesos y 79 % tenían glucemia y nivel de lípidos, normales. Craig y otros38 reportaron igual frecuencia en hombres que en mujeres y que sólo 40 % de los pacientes eran obesos, 15 % diabéticos y 20 % dislipidémicos.
Los 3 criterios propuestos por Powell y otros39 para el diagnóstico de EGNAH son:
- Un cuadro histológico de esteatohepatitis.
- La evidencia convincente de mínimo o ningún consumo del alcohol (< 40 g/sem).
- La ausencia de evidencias serológicas de hepatitis virales.
Aunque estos criterios se usan ampliamente en la práctica clínica, cada criterio tiene sus limitaciones específicas. El término histológico de esteatohepatitis utilizado por Powell39 limita el diagnóstico a una sola forma histológica de la enfermedad, deja fuera de este criterio a la estatosis simple y a los grados más avanzados de la enfermedad, además excluye la posibilidad de realizar el diagnóstico sin realizaciar la biopsia hepática. Respecto a los límites precisos en términos de la cantidad consumida de alcohol necesaria para el diagnóstico de EGNAH, se han planteado diferentes límites y se han intentado marcadores directos del consumo de alcohol sin una definición clara del problema. La presencia de marcadores virales positivos se creía originalmente que constituía un criterio de exclusión para el diagnóstico de EGNAH, pero varios investigadores han visto en pacientes con hepatitis C40 una histología que evidencia una esteatohepatitis clásica, en lugar de tener predominantemente linfocitos que infiltran la región periportal, con moderada esteatosis como es frecuente en la hepatitis C. En tales casos, la presencia de 2 diagnósticos (hepatitis C y EGNAH) puede ser considerada.41 Aunque por consenso se debe excluir la presencia de hepatitis virales B, C, G y D.
Hasta dónde investigar
La biopsia hepática es el único examen que permite el diagnóstico exacto de EGNAH, pero es impracticable de rutina. No hay otro examen cuyo resultado pueda ser homologable al de la biopsia. En la práctica clínica, el diagnóstico de EGNAH debe sospecharse por la suma de factores y marcadores de riesgo. Generalmente, el paciente consulta al clínico por el resultado de un ultrasonido que sugiere la infiltración grasa del hígado o por la elevación de las transaminansas, asociada o no a manifestaciones clínicas. Aunque teniendo los elementos clínicos, imagenológicos, las pruebas de función hepática con el índice ASAT/ALAT menor a uno se puede prescindir de la biopsia hepática, iniciar las medidas terapéuticas y reservar la biopsia para los casos de alta sospecha de progresión de la enfermedad, como menores de 45 años, obesos o en mayores de 45 años, diabéticos, obesos, con dislipidemias y el índice ASAT/ALAT menor que 2 y mayor que 1 o cuando se sospecha otro diagnóstico etiológico o existan estigmas periféricos de daño hepático crónico y de manifestaciones clínicas secundarias a cirrosis hepática.42,43
Factores de riesgo y redefinición de criterios de mal pronóstico
Las condiciones predisponentes son la obesidad, la diabetes tipo 2, el sexo femenino, la insulinorresistencia, el bypass yeyunoileal, la dislipidemia tipo IV y la desnutrición, pero a partir de la anterior reflexión acerca de la ruptura con los estereotipos clínicos tradicionales, se deben replantear los factores de riesgo para la progresión de la enfermedad ya que algunos estudios indican que la EGNAH es la causa más importante de cirrosis hepática criptogénica,44,45 que 3 % de los pacientes que llegan al trasplante hepático son por su causa46 y que después de este, recidiva el daño en un gran porcentaje de los casos.
Fernández-Salazar y otros47 publicaron un estudio con 53 sujetos que tenían hepatitis crónica C en el que encontraron esteatosis hepática en 50 % de estos sujetos y que los factores asociados, independientemente con su presencia fueron la sobrecarga de hierro y la infección por el genotipo 3 del virus de la hepatitis C. Actualmente, se ha prestado gran interés al papel que desempeña el depósito de hierro como factor de riesgo de la EGNAH.
Dentro de los factores de mal pronóstico se encuentran 4 que pueden actuar combinados o solos (edad mayor que 45 años, obesidad, diabetes mellitus tipo 2 y dislipidemias)48 más el índice ASAT/ALAT mayor que 1. Además de una biopsia hepática al inicio del diagnóstico con lesiones intensas. Y se deben tener otros factores en cuenta como son: la coexistencia con hepatitis C, lesiones de siderosis hepática, niveles muy altos de ALAT y de γ-glutariltranspeptidasa. Otros autores incluyen los niveles elevados de péptido C49 y la presencia del síndrome metabólico.
Enfoque terapéutico
El tratamiento debe tener 4 objetivos fundamentales: disminuir el flujo de ácidos grasos libres al hígado, proteger al hepatocito de mecanismos oxidativos, evitar tóxicos conocidos que dañen al hígado y tratar los factores condicionantes o asociados a la EGNAH.
Para reducir el flujo de ácidos grasos libres al hígado se recomienda reducir de peso corporal de forma gradual (de 0,5 a 1 kg/sem) y la administración de agentes hipolipemiantes, en particular los derivados del ácido fíbrico.50 En los diabéticos y en la insulinorresistencia, la administración de drogas que mejoren la resistencia a la insulina.
Para proteger al hepatocito del estrés oxidativo se han propuesto la vitamina E y el ácido ursodesoxicólico50 del cual se recomienda una dosis no superior a 10-15 mg/kg de peso por día. También la lecitina, el selenio y la betaina son otras sustancias usadas, pero no hay evidencias definitivas sobre su efectividad. En nuestro mercado, no existe ningún citoprotector hepático disponible. Investigadores del Departamento de Farmacología del Centro Nacional de Investigaciones Científicas (CENIC), realizaron un estudio del efecto antioxidante de un extracto etanólico de propóleo rojo cubano en roedores y reportaron mejoría histológica de la esteatosis inducida con tetracloruro de carbono y reducción de los niveles de ALAT.51
Para evitar tóxicos conocidos que dañen al hígado se debe recomendar a los pacientes tratados que no consuman alcohol, ni drogas hepatotóxicas y tratar los factores condicionantes o asociados a la EGNAH (figura).52
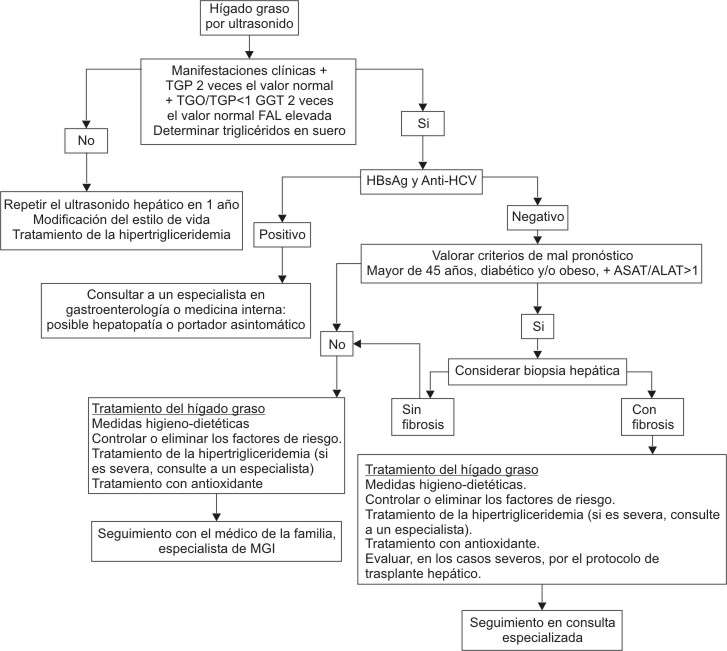
Fig. Algoritmo del tratamiento y seguimiento.
Summary
Fatty liver. Diagnostic and therapeutic approach
The diagnostic and therapeutic approach on fatty liver, proposed in this review, was made to meet the needs of the practitioner. A theoretical disquisition on the terminological problem of this syndrome was stated, and the common pathogenic mechanisms in the different clinical conditions predisposing to suffer from this health problem, that may appear as a simple hepatic steatosis, but that may also be the cause of liver cirrhosis leading the patient to a liver transplantation, were approached. It was considered the importance of hefting the sum of the factors predisposing the disease, the imaging studies, and the risk markers of the development of fibrosis before indicating the liver biopsy. The treatment is intended to reduce the flow of fatty acids to the liver, to protect the hepatocyte from oxidative mechanisms, to avoid known toxic agents damaging the liver, and to treat the conditioning or associated factors. An algorithm of care and follow-up of these patients was suggested.
Key words: Non-alcoholic fatty liver disease, simple esteatosis, esteatohepatitis, metabolic disease.
Referencias bibliográficas
1. Nonomura A, Mizukami Y, Unoura M, Kobayashi K, Takeda Y, Takeda R. Clinicopathologic study of alcohol-like liver disease in non-alcoholics; nonalcoholic steatohepatitis and fibrosis. Gastroenterol Jpn. 1992;27:521-8.
2. Sheth SG, Gordon FD, Chopra S. Nonalcoholic Steatohepatitis. Ann Intern Med. 1997;126(2):137-45.
3. Medina J, Fernández-Salazar LI, García-Buey L, Moreno-Otero R. Approach to the patogénesis and treatment of nonalcoholic steatohepatitis. Diabetes Care. 2004;27:2057 - 66.
4. Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346(16):1221-31.
5. Ludwig J, Viggiano TR, McGill DB, Oh Bj. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. 1980;55(7):434-8 .
6. Cohen H, González M, Ramírez M. Etiología y diagnóstico de la esteatosis hepática. Rev Med Uruguay. 1997;13:4-11.
7. Sanyal AJ, Campbell-Sargent C, Mirshani K. Nonalcoholic steatohepatitis: Association of Insulin resistance. Gastroenterology. 2001;107:1183-92.
8. Kotish A, Diehl AM. Animal models of steatosis. Semi Liver Dis. 2001;21:89-104.
9. Younossi ZM, Diehl AM, Ong JP. Nonalcoholic fatty liver disease: an agenda for clinical research. Hepatology. 2002;35:746-52.
10. Youssef W, McCullogh AJ. Diabetes Mellitus, Obesity, and hepatic Steatosis. Sem Gastrointest Dis. 2002;13(1):17- 30.
11. Ma X , Li Z . Pathogenesis of nonalcoholic steatohepatitis. Chin J Dig Dis. 2006;7(1):7-11.
12. Pessayre D, Berson A, Fromenty B, Mansoure A. Mitocondria in steatohepatitis. Sem Liver Dis. 2001;21:57-69.
13. Pessayre D, Mansouri A, Fromenty B. Nonalcoholic steatosis and steatohepatitis V. Mitochondrial dysfunction in steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2002;282:G193-9.
14. Pérez-Carreras M, Hoyo P, Martín MA, Rubio JC, Martín A, Castellano G, et al. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology. 2003;38:999-1007.
15. Chitturi S, Farrell GC. Etiopathogenesis of nonalcoholic steatohepatitis. Semin Liver Dis. 2001;21:27- 41.
16. Washington K, Wright K, Shyr Y. Hepatic stellate cell activation in nonalcoholic steatohepatitis and fatty liver. Hum Pathol. 2000;31:822-8.
17. Clouston AD, Jonsson JR, Purdie DM, Macdonald GA, Pandeya N, Shorthouse C, et al. Steatosis and chronic hepatitis C: analysis of fibrosis and stellate cell activation. J Hepatol. 2001;34:314-20.
18. Valenti L, Fracanzani Al, dongiovanni P, Santorelli G, Branchi A. Tumor necrosis factor alpha promoter polymorphisms and insulin resitance in non alcoholic fatty liver disease. Gastroenterology. 2002;122:274-80.
19. Bernard S, Touzet S, Personne I, Lapras V, Bondon PV, Berthezene F, et al. Association between microsomal triglyceride transfer protein gene polymorphism and the biological features of liver steatosis in patients with type II diabetes. Diabetologia. 2000;43:995-9.
20. Namikawa C, Shu-Ping Z, Wyselaar JR, Nozaki Y, Nemoto Y, Ono M, et al. Polymorphisms of microsomal triglyceride transfer protein gene and manganese superoxide dismutase gene in non-alcoholic steatohepatitis. J Hepatol. 2004;40:781-6.
21. Wigg AJ, Roberts.Thomson IC, Dymock RB. The role of small intestinal bacterial overgrowth, intestinal pereabiliity, endotoxaemia and tumour necrosis factor alpha in the pathogenesis of nonalcoholic steatohepatitis. Gut. 2001;48:206-11.
22. Tilg H, Diehl AM. Cytokines in alcoholic and nonalcoholic steatohepatitis. N Engl J Med. 2000;343:1467-76
23. Marchesini G, Brizi M, Morselli-Labate A.M. Bianchi G, Bugianesi E, McCullough A.J. et al. Association of Non alcoholic fatty liver disease with insulin resistance. Am J Med. 1999;17:80-89.
24. Kral JG, Shaffner F, Pierson RN Jr, Wang J. Body fat topography as an independent predictor of fatty liver. Metabolism. 1993;42:548-51.
25. Diehl AM, Goodman Z, Isask KG. Alcohol-like liver disease in nonalcoholics. a clinical and histologic comparison with alcohol induced liver injury. Gastroenterology. 1988;95:1056-62.
26. Lee RG. Non-alcoholic steatohepatitis: A study of 49 patients. Hum Pathol. 1989;20:594-8.
27. Bacon BR, Farahvash MJ, Janney CG, Neuschwander-Tetri BA. Nonalcoholic steatohepatitis: an expanded clinical entity. Gastroenterology. 1994;107:1103-9.
28. Brunt EM. Nonalcoholic Steatohepatitis: definition and Pathology. Sem Liver Dis. 2001;21(1):3-16.
29. Teli MR, James OF, Burt AD, Bennett MK, Day CP. The natural history of non-alcoholic fatty liver: a follow-up study. Hepatology. 1995;22:1714-9.
30. Friedman LS, Dienstag JL, Watkins E. Evaluation of blood donors with elevated serum alanine aminotransferase. Ann Intern Med. 1987;107:137-44.
31. Clain DJ, Lefkowitch JH. Fatty liver disease in morbid obesity. Gastroenterol Clin North Am. 1987;16(2):239-52.
32. Sorbi D, Boynton J, Lindor KD. The ratio of aspartate aminotransferase to alanine: potencial value in differentiating nonalcoholic steatohepatitis from alcoholic liver disease. Am J Gastroenterol. 1999;94(4):1018-22.
33. Rantala. AO, Lilja M, Kauma H, Savolainen MJ, Reunanam.A, Kesaniemi YA. Gamma-glutamyl transpeptidase and the metabolic syndrome. J Int Med 2000;248:230-38.
34. Evan S, Siegelman, Rosen MA. Imaging of Hepatic Seteatosis. Sem Liver Dis. 2001;21(1):71- 80.
35. Clark J, Brancuti F, Diehl A. Nonalcoholic fatty liver disease. Gastroenterology. 2002;122:1649-57.
36. Ratziu V, Giral P, Charlotte F. Liver fibrosis in overweight patients. Gastroenterology. 2000;118:1117-23.
37. Bacon BR, Farahvash MJ, Janney. Nonalcoholic Steatohepatitis: an expanded clinical entity. Gastroenterol. 1994,107:1103- 5 .
38. Craig RM, Neumann T, Jeejeebhoy KN, Yokoo H. Severe hepatocelular reaction resembling alcoholic hepatitis with cirrosis after massive small bowel resection and prolonged total parenteral nutrition . Gastroenterology. 1980;79:131-7.
39. Powell EE, Cooksley WG, Hanson R, Searle J, Halliday JW, Powell LW. The natural history of nonalcoholic steatohepatitis: a follow-up study of forty two patients for up to 21 years. Hepatology. 1990;11:74-80.
40. Rogers DW, Lee CH, Pound DC, Kumar S, Cummings OW, Lumeng L. Hepatitis C virus does not cause non-alcoholic steatohepatitis. Dig Dis Scl. 1992;37:1644-7.
41. Sanyal AJ , Contos MJ , Sterling RK , Luketic VA , Shiffman ML , Stravitz RT et al. Nonalcoholic fatty liver disease in patients with hepatitis C is associated with features of the metabolic syndrome. Am J Gastroenterol . 2003;98(9):2064-71.
42. Matteoni C, Younossi ZM, Gramlich T. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413-9.
43. Angulo P, Keach JC, Butts KP. Independent predictors of liver fibrosis in pacients with nonalcoholic steatohepatitis. Hepatology. 1999;30:421-9.
44. Caldwell SH, Oelsner D, Iezzoni JC. Cryptogenic cirrhosis: Clinical characterization and risk factors for underlying disease. Hepatology. 1999;29:664-9.
45. Poonawala A, Nair SP, Thuluvath PJ. Prevalence of obesity and diabetes in patients with cryptogenic cirrhosis: A case- control study. Hepatology. 1999;32:6889-99.
46. Baiocchi L, Tisone G, Palmieri G, Rapicetta M, Pisani F, Orlando G, et al. Hepatic steatosis: a specific sign of hepatitis C reinfection after liver transplantation. Liver Transpl Surg. 1998;4:441-7.
47. Fernández-Salazar LI, Álvarez-Gago T, Aller de la Fuente R , Orduña-Domingo A, Arranz-Santos T, de la Calle-Valverde F , et al. Iron overload and genotype 3 are associated with liver steatosis in chronic hepatitis C. Rev Esp Enferm Dig. 2004; 96 (12): 818-28
48. López D, Casal E, Barbado H, Gil G, Rodríguez M, larrauri J et al. Esteatohepatitis no alcoholica, el enigma de una mala evolución. An Med Interna. 2005 Feb;22(2):85-7.
49. Dixon JB, Bhathal PS, O Brian PE. Non – alcoholic steatohepatitis and liver fibrosis in the severely obese.Gastroenterology. 2001;121:91–100.
50. Laurin J, Lindor KD, Crippin JS, Gossard A, Gores GJ, Ludwig J, et al. Ursodeoxycholic acid or clofibrate in the treatment of non-alcohol-induced steatohepatitis: a pilot study. Hepatology. 1996; 23:1464-7.
51. Rodríguez S, Ancheta O, Ramírez D, González A, Merino N, González R. Efecto protector del propóleo rojo cubano ante el daño agudo inducido en hepatocitos de roedores . Revista CENIC Ciencias Biológicas 1998;29(2):69-72.
52. Pérez-Aguilar F, Benlloch S, Berenguer M, Beltrán B, Berenguer J. Non-alcoholic steatohepatitis: physiopathological, clinical and therapeutic implications. Rev Esp Enferm Dig. 2004;96(9):628-48
Recibido: 28 de julio de 2006. Aprobado: 26 de octubre de 2006.
Dr. Alfredo Herrera González. . Hospital Clinicoquirúrgico "Hermanos Ameijeiras" San Lázaro No. 701 entre Belascoaín y Marqués González, Centro Habana, Ciudad de La Habana , Cuba. CP 10300. Correo electrónico: aaerrez@infomed.sld.cu
1Especialista I Grado en Medicina Interna. Profesor. Hospital Clinicoquirúrgico "Hermanos Ameijeiras."
2Especialista II Grado en Medicina Interna. Profesor. Hospital Clinicoquirúrgico "Hermanos Ameijeiras."
3 Especialista II Grado en Gastroenterología. Profesor. Instituto de Gastroenterología.
4 Especialista II Grado. Profesora de Imageneología. Hospital Clinicoquirúrgico "Hermanos Ameijeiras."

