










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La holoprosencefalia (HPE) constituye una malformación cerebral en la que el prosencéfalo o el prosencéfalo embrionario no se divide en dos lóbulos separados.1 Presenta una incidencia de 1:250 durante la etapa prenatal y ocurre en uno de cada 16 000 nacidos vivos.2) Esta anomalía resulta la malformación más común del prosencéfalo en los seres humanos, es una anomalía estructural del cerebro resultante de una división del prosencéfalo fallida o incompleta en la tercera a cuarta semana de gestación. El prosencéfalo (prosencéfalo embrionario) se divide de forma incompleta en los hemisferios derecho e izquierdo, las estructuras cerebrales profundas, y los bulbos y tractos olfatorios y ópticos.3
En la literatura existe una amplia variabilidad clínica, según el subtipo de HPE. La letalidad posnatal temprana resulta común; sin embargo, se encuentra supervivencia en la infancia e, incluso, en la edad adulta.4) Esta enfermedad da lugar a una amplia gama de consecuencias fenotípicas para el cerebro y el rostro de los seres humanos como consecuencia directa de agresiones genéticas o ambientales determinadas que interrumpen la especificación de la línea media del prosencéfalo naciente.5
La HPE se asocia con mutaciones heterocigóticas en los componentes de la vía Nodal y la proteína sonic hedgehog (SHH), encargada de la regulación de la organogénesis de los vertebrados, como el crecimiento de las falanges de las extremidades y la organización del cerebro. La presentación clínica es muy variable, como se comentaba anteriormente, y muchos portadores de mutaciones no se ven afectados.6 La mayoría de los casos son esporádicos; sin embargo, las anomalías cromosómicas presentes se ubican entre el 25 y el 50 % de las personas con HPE; la trisomía 13 comprende el 75 %; la triploidía, el 20 %; y la trisomía, entre el 18 y 1 a 2 %.7) En el mundo la prevalencia registrada de HPE en neonatos nacidos vivos es de uno por 10 000.1,5
La HPE ocurre entre los 18 y 28 días de gestación,3,8 lo que indica que se trata de un trastorno de la gastrulación; es una anomalía estructural rara del cerebro y se asocia con deterioro neurológico y dimorfismo facial.1,8,9) Autores como Demyer y Zeman,10) en 1963, sugirieron que esto se debía a un defecto en la inducción ventral y al patrón del tubo neural rostral por el mesénquima precordial. Como la inducción ventral está relacionada con el desarrollo facial, muchos casos de HPE también muestran anomalías craneofaciales, lo que lleva a la denominada “secuencia de holoprosencefalia”.2
La clasificación más utilizada de esta enfermedad resulta la propuesta por Demyer y Zeman,10) también en 1963, que destaca tres tipos de HPE: alobar, semilobar y lobar, según el grado de separación de los hemisferios y la presencia o ausencia de la fisura interhemisférica.3,10) Esta clasificación también resulta útil desde un punto de vista pronóstico, ya que ofrece información sobre la gravedad y el impacto neurológico de la malformación. En la forma más grave, HPE alobar, existe una falta total de escisión de la corteza, mientras que, en la forma más leve, HPE lobar, solo una parte de los lóbulos frontales y los cuernos ventriculares resultan fusionados.8Barkovich y Quint,11 en 1993, describieron una nueva variante de HPE denominada variante “interhemisférica media” (MIH) o sintelencefalia, en la que la principal característica estaba dada por la existencia de un grado de fusión interhemisférica media.11
El primer diagnóstico prenatal de HPE lo realizaron Kurtz y otros12) en 1980. Desde entonces, algunos estudios e informes de casos describieron los diferentes hallazgos ecográficos de HPE.
En Cuba, según datos procedentes del Registro Cubano de Malformaciones Congénitas (RECUMAC) durante sus primeros diez años de trabajo, comprendidos entre abril de 1985 y diciembre de 1994, se diagnosticaron 11 casos con esta enfermedad, los cuales representaron una frecuencia de 0,25 por cada 10 000 nacidos vivos y una tasa de 3,8 % en relación con el total de casos (288) con malformaciones congénitas diagnosticados durante ese período.13
El objetivo de este trabajo fue describir, en un paciente neonato, las manifestaciones clínicas en enfermos con holoprosencefalia, conducta clínica y terapéutica.
Presentación del caso
Paciente: hijo de padres no consanguíneos, madre de 35 años de edad con antecedente de cervicitis y gestorragia en la segunda mitad del embarazo, y antecedentes familiares de diabetes mellitus y cardiopatía.
El parto se produjo a término a las 37 semanas, distócico por cesárea secundaria a un hematoma retroplacentario. Se obtuvo un recién nacido del sexo masculino con presentación pelviana que no precisó reanimación, con un Apgar 9/9 al nacer y peso de 3380 g, procedente de área de salud urbana y nacido en el Hospital Materno Sur “Mariana Grajales Coello”, de Santiago de Cuba. Al nacer se apreciaron rasgos dismórficos en el neonato, principalmente cráneo-facial, y a los pocos minutos comenzó con cuadro de dificultad respiratoria, además de hiposaturación.
Al examen físico se constató lo siguiente:
Frecuencia respiratoria de 65 por min.
Frecuencia cardíaca de 135 latidos por min.
Tensión arterial media de 50 mmHg.
Además, se precisó una saturación de oxígeno de 84 % por pulsioxímetro.
A nivel de la cabeza (figs. 1 A, B y C, y 2 A y B):
Cara: hipoplasia medio facial, hipoplasia del puente nasal, protrusión de los glóbulos oculares, hipoplasia de órbita, implantación baja de las orejas, orificio nasal pequeño, implantación baja del cabello que se une con la cola de las cejas, micrognatia, paladar duro pronunciado en el rafe medio, con insuficiencia velo palatina.
Cráneo: dolicocefalia, fontanela anterior normotensa, circunferencia cefálica de 35 cm.
Cuello: corto con pliegue nucal (fig. 1 B).
Tórax: circunferencia torácica 33 cm.
Talla de 50 cm.
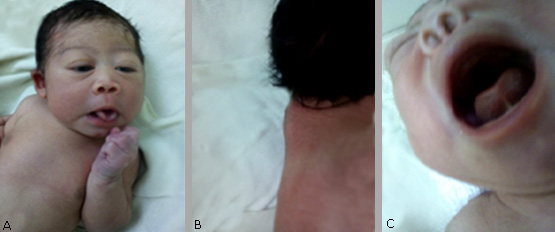
Fig. 1 A: se observa hipoplasia medio facial, hipoplasia del puente nasal, protrusión de los glóbulos oculares, hipoplasia de órbita, implantación baja del cabello que se une con la cola de las cejas, y micrognatia. B: Se observa cuello corto y pliegue nucal. C: paladar duro pronunciado en el rafe medio y orificio nasal pequeño.
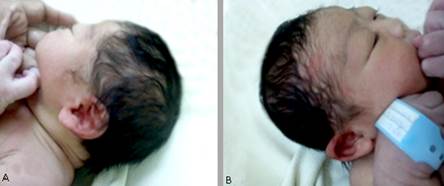
Fig. 2 A: se observa dolicocefalia, implantación baja de las orejas, puente nasal aplanado. B: implantación baja del cabello que se une con la cola de las cejas.
Ingresa en la unidad de cuidados intensivos neonatales (UCIN) inmediatamente, luego de ser reportado de grave, bajo monitorización con equipo DOCTUS VIII de forma no invasiva, con oxigenoterapia a 5 l/min, por la clínica y tener gases arteriales en acidosis respiratoria con hipoxemia en un comienzo, que se retiró paulatinamente luego de obtener adecuados índices de oxigenación. Presentó tiraje subcostal, además de obstrucción nasal; sin embargo, no manifestó alteraciones electrolíticas y el resto de los estudios de laboratorio, como glicemia, coagulograma y hemograma completo, estuvo dentro de los límites normales. También se realizó radiografía de tórax, que se informó sin alteraciones. Evolutivamente se constató que el neonato durante la alimentación y cuando cerraba la boca, comenzaba con dificultad respiratoria; mientras no se mantuvo en esta condición no mostraba alteración del patrón respiratorio.
Se decidió su valoración por otorrinolaringología y se realizó una rinoscopia anterior, además de pasar sonda de aspiración para explorar permeabilidad de ambas fosas nasales y se decidió colocar cánula de Gueder, y valorar por maxilofacial y genética. Estos especialistas indicaron otros estudios como ecocardiograma, el cual informó comunicación interauricular (CIA) de 1,5 mm, que no justificaba el cuadro, y ultrasonido abdominal sin alteraciones clínicas.
Los genetistas indicaron un ultrasonido transfontanelar, el cual informó una disgenesia del cuerpo calloso sin delimitar su porción distal, no se limitaban las astas posteriores de los ventrículos laterales, persistencia del cavus vergae de 6 mm, ventrículos laterales dilatados, además de la cisterna magna de 12 mm, y cuarto ventrículo de 9 mm. Por estos resultados se sugirió realizar tomografía axial computarizada (TAC) de cráneo para confirmar el diagnóstico de holoprosencefalia a los cinco días de nacido, la cual informó ventrículos laterales dilatados en 13 mm, cisterna magna dilatada; en cortes altos se observó imagen hiperdensa de forma alargada de 51 Hounsfield (UH) (fig. 3).
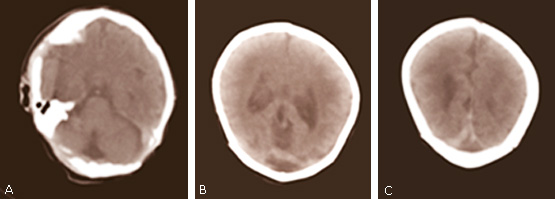
Fig. 3 Imágenes de la tomografía axial computarizada de cráneo, monocorte Shimasu del caso. A: se observa cisterna magna dilatada; B: ventrículos laterales dilatados; C: imagen hiperdensa de forma alargada.
Después de la colocación de la cánula de Gueder en este recién nacido, se observó una evolución favorable: tolerancia de la alimentación sin presentar más dificultad al respirar. Se reportó en estado de cuidado y alta evolutiva con seguimiento por otorrinolaringología, maxilofacial, consulta de neurodesarrollo, neuropediatría, fisiatría, genética, para recibir tratamiento rehabilitador.
Discusión
El diagnóstico de la holoprosencefalia se realiza mediante el estudio prenatal, por ecografía, más factible en el primer trimestre, con el hallazgo de fusión de los cuernos anteriores de los ventrículos laterales y signo de mariposa.3 Sin embargo, este caso no tuvo diagnóstico en esta etapa; se constató esta enfermedad en el período posnatal por los rasgos fenotípicos al examen físico. Autores como Kagan y otros,14) en 2010, señalaron posible presencia de las malformaciones faciales de línea media como hipotelorismo, probosis, alteraciones diversas de nariz, labio y paladar, al igual que en el paciente presentado. Además, el 50 % de los casos muestra anomalías extracraneales: mielomeningocele, displasia renal, cardiopatías y polidactilia.3
En este caso, con el ultrasonido y la TAC se confirmó la presencia de una holoprosencefalia de tipo lobar.
Esta malformación se presenta debido a una falla en el desarrollo del cerebro anterior, probablemente por error en el crecimiento de la porción anterior final del tubo neural. Las malformaciones oculares son las de mayor incidencia, seguidas por las malformaciones nasales, del oído, la boca y el paladar.4
Entre los supervivientes, el resultado a largo plazo resulta desfavorable y depende del tipo y la gravedad de la enfermedad, así como de las anomalías asociadas. Los problemas neurológicos, como la parálisis cerebral y las crisis epilépticas se presentan de forma reiterada. Algunos pacientes manifiestan espasticidad, movimientos atetósicos o retraso mental.15
Favorablemente, el paciente egresó vivo, aunque no estuvo exento de que en su desarrollo psicomotor, evolutivamente, presentara alguna de las complicaciones mencionadas. Por tal motivo, se indicó seguimiento por consulta de neurodesarrollo, además de las especialidades involucradas en esta enfermedad para una mejor evolución.
La letalidad posnatal temprana es común; sin embargo, una fracción significativa de los recién nacidos diagnosticados con HPE sobreviven hasta la infancia e, incluso, hasta la edad adulta.16
Existen factores predisponentes que influyen en la presencia de esta enfermedad. Entre los factores ambientales se encuentra la diabetes materna mal controlada, y se sospecha de otros, como la ingestión de alcohol, infecciones prenatales como el complejo TORCH (toxoplasmosis, rubeola, citomegalovirus, herpes simple, entre otras), ingestión materna de salicilatos, altas dosis de anticonceptivos, quinina, algunos medicamentos como el ácido 3-5 retinoico, estatinas, cortisona o irradiación.15,17) En el caso presentado se recogió el antecedente familiar de diabetes mellitus, enfermedad que puede aumentar 200 veces el riesgo.3,8,18
Se concluye que, en la holoprosencefalia, el diagnóstico posnatal se puede realizar mediante las características fenotípicas, las malformaciones faciales y los estudios neuroimagenológicos como el ultrasonido transfontanelar y la tomografía axial computarizada de cráneo. Los pacientes deben evaluarse y seguirse en la evolución por un equipo multidisciplinario de especialidades como otorrinolaringología, máxilo-facial, neuropediatría, consulta de neurodesarrollo, genética, fisiatría e imagenología.

