










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Las porfirias hepáticas agudas son un trastorno genético causado por la actividad irregular de las enzimas en la vía metabólica del grupo hemo; importante para la síntesis de hemoglobina, mioglobina, citocromo p450, entre otros.1,2 Estos trastornos se clasifican en agudas y no agudas; las primeras se consideran de alto riesgo de muerte.3,4) La presentación aguda se manifiesta en ataques intermitentes súbitos con síntomas neuroviscerales, lo cual aumenta la necesidad de hospitalización en la unidad de cuidados intensivos (UCI).5,6
Debido a que la sintomatología es inespecífica, el diagnóstico puede ser tardío, lo cual repercute en el tratamiento oportuno.7 La primera alternativa diagnóstica es el uso de pruebas bioquímicas (orina - sangre - heces), aunque las pruebas genéticas son consideradas la mejor opción diagnóstica.4,8 El tratamiento aprobado de la crisis aguda, es iniciar soporte nutricional rico en carbohidratos, infusiones intravenosas de glucosa y hemina.9 La terapia génica se encuentra como nueva opción de tratamiento y mejora la calidad de vida.10,11
Se realizó una revisión bibliográfica a través de bases de datos electrónicas PubMed, Medline, ScienceDirect y SciELO, con los términos “acute hepatic porphyria”, “heme” “hemina”. Se incluyeron reportes de caso, revisiones bibliográficas y serie de casos en inglés o español, desde enero del 2015 a diciembre del 2020, los cuales describen porfirias agudas hepáticas, se seleccionaron 59 artículos.
El objetivo de esta revisión es actualizar sobre las alternativas diagnósticas y terapéuticas para las porfirias hepáticas agudas en adultos.
DESARROLLO
Definición
Según la American Porphyria Foundation, las porfirias agudas son un grupo de trastornos metabólicos y clínicos, causados por una deficiencia enzimática en la biosíntesis del grupo hemo, de tal manera que aumentan los productos bioquímicos tóxicos (ácido delta amino levulínico (ALA) y porfobilinógeno (PBG)) en sangre.5,12 Esto desencadena un ataque agudo neurovisceral, que compromete la vida del paciente.12 Existen 4 tipos de porfirias hepáticas agudas: porfiria aguda intermitente (PAI); coproporfiria hepática (CPH); porfiria variegata (PV) y porfiria por deficiencia ALA deshidratasa (ALAD-P).1,13
Epidemiología
La prevalencia global de las porfirias hepáticas aguda es de 5 por cada 100 000 habitantes.14 Se presenta después de la pubertad y antes de la menopausia, con excepción de la ALAD-P, que aparece de manera temprana en la infancia.15 La presencia activa es baja, debido a que el 90 % de los pacientes permanecen asintomáticos de por vida.16 En Europa, la incidencia anual es de 0,13 por millón de habitantes, excepto en Suecia, que se ha calculado de 1 por cada 1000 habitantes, debido a migración genética.17,18 Hasta la fecha en Latinoamérica, se desconocen datos epidemiológicos acerca de estos trastornos.
La PAI es la porfiria hepática aguda más común, con una prevalencia de 5,4 por millón de habitantes en los países de Europa.19 La PV y CPH tienen una prevalencia de 1 por cada 30 000 y 50 000 habitantes respectivamente.20,21 En la ALAD-P, hasta la fecha, solo se han documentado 6 casos, que presentaron síntomas iniciales a los 14 años.22 Este bajo número se explica debido a que el 50 % de la actividad del ALAD es normal, razón por la cual los pacientes permanece asintomático durante toda la vida.22
Síntesis de hemo y regulación
La biosíntesis de hemo consiste en 8 reacciones enzimáticas que ocurren en el citosol y las mitocondrias (Fig. 1).23,24 Del 15 % al 20 % se forma a nivel hepático, mediado a través de la 5-aminolevulinato sintasa 1 (ALAS1); el 80 % restante es producido por la médula ósea, regulado por la ALAS2, que controla la velocidad de producción.25,26
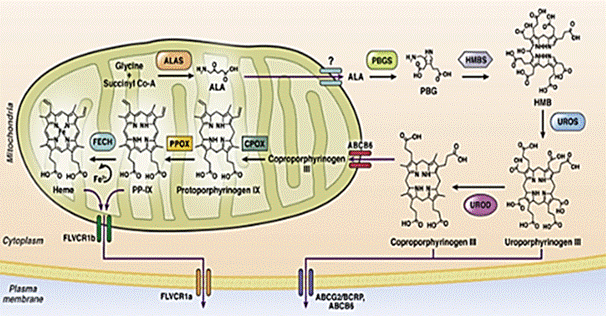
Las porfirinas son tetrapirroles cíclicos conectados por puentes de metano, de carbono y un sitio central de metal.23 El hemo a concentraciones fisiológicamente alcanzables, disminuye la estabilidad de ARN mensajero ALAS1 (ARNm) y la velocidad de formación de ALA y PBG.18 A nivel hepático aumenta la absorción mitocondrial de pre-ALAS1 y la descomposición citosólica de la enzima.25 La actividad de ALAS1 también está regulada negativamente por niveles altos de glucosa, debido a la modulación del coactivador del receptor 1 activado por el proliferador de peroxisomas (PCG-1).26 En la médula ósea el ALAS2 está bajo la regulación de factores de transcripción eritroides, como el factor de unión GATA-1 y el regulador de hierro.26
Tipos de porfirias hepáticas agudas
Las porfirias se clasifican en hepáticas o cutáneas, según el sitio primario de sobreproducción, la enzima deficiente y la acumulación de precursores, como se observa en la tabla 1.7,15,17 La PAI es causada por un defecto del gen HMBS que abarca 10 kb en el cromosoma 11q23; existen hasta 400 mutaciones diferentes.27 La CPH se debe al defecto del gen CPg-O-CPOX del cromosoma 1 región 3q12.1 con 130 mutaciones identificadas.20,28
La PV es consecuencia de cambios defectuosos del gen PPOX, que se encuentra en el cromosoma 1 de la región R59W.22,28 La deficiencia de ALAD-P se debe a mutaciones en el gen de la enzima de ALAD codificado por el cromosoma 9q34 con un tamaño de 34 kb.22 Independientemente del tipo de porfiria hepática aguda, el 90 % de los portadores genéticos confirmados no experimentan una crisis aguda en su vida.29
Tabla 1 Presentación clínica y enzimática de las porfirias agudas y cutáneas
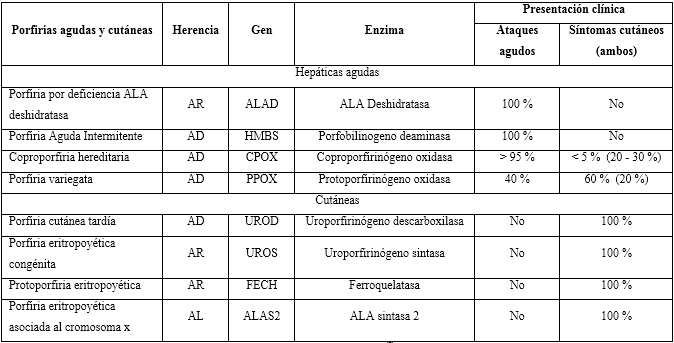
Fuente: Stein y otros.7
AR: Autosómico recesivo; AD: Autosómico dominante, AL: Ligado cromosoma X
Etiología
Aunque su etiología aún no es clara, existen factores desencadenantes que conllevan a una crisis aguda.5 Estos incluyen, sustancias psicoactivas, hormonas esteroideas, estrés, procedimientos quirúrgicos, alcohol, infecciones y respuestas endocrinas (ciclo menstrual, cambios en fase lútea y aumento de hormonas sexuales en la pubertad).8,30 Al evitar estos factores, el 50 % de la actividad enzimática se reduce y mantiene la normalidad.3,15 Según El Centro Noruego de Porfiria y la Red Europea de Porfiria, fármacos antiepilépticos (barbitúricos, diazepam, ácido valproico, fenobarbital, fenitoína, carbamazepina), anestésicos (tiopental, ketamina), antihipertensivos (hidralazina, nifedipino) y antibióticos (rifampicina, eritromicina, nitrofurantoína, trimetoprim sulfametoxazol, cloranfenicol, sulfamidas), entre otros, son precipitantes de ataques agudos.9,30 Esto induce la actividad de ALAS1, ALAS2 y el citocromo p450, que genera un incremento en la producción de productos de porfirinas hepáticas.2,25
El envenenamiento agudo por plomo y la tirosinemia hereditaria tipo I, aumenta la producción de ALA.15 Estos inactivan a la ALA-D en su sitio activo, causan un aumento de la ALA en sangre y desencadenan un ataque agudo.25
Fisiopatología
La aparición del ataque agudo resulta del defecto genético/enzimático de la biosíntesis del hemo y los factores precipitantes que inducen la expresión del gen ALAS1 en el hígado.17 Este aumenta los precursores PBG-ALA, responsables de los síntomas neurológicos y gastrointestinales.31 Estas respuestas actualmente son poco comprendidas, debido a la ausencia síntomas en la mayoría de los casos, sin embargo, existen teorías aisladas que explican el mecanismo de daño.2,32
Hiperactividad autónoma
La neurodegeneración es un proceso complejo que conduce a la pérdida progresiva y selectiva de neuronas del sistema autónomo.33 Esto conlleva a hiperactividad autónoma, factor causal de vasoespasmo, taquicardia, náuseas e hipertensión.3,33) Esta respuesta en el intestino, exacerba el vasoespasmo y la dilatación, que conduce a lesiones ganglionares responsables del dolor abdominal.2)
En el sistema nervioso central (SNC) la hiperactividad autónoma disminuye la producción de óxido nítrico.34 Esto genera una vasoconstricción, que excede el límite de autorregulación y causa hipertensión grave.9,32 La hipoperfusión aumenta la presión hidrostática capilar que favorece la extravasación intersticial y edema vasogénico en la sustancia blanca cerebral, lleva a un síndrome de encefalopatía reversible posterior (SEPR).9 Cuando la hipertensión aguda no excede los límites de autorregulación cerebral, presenta disfunción endotelial, ruptura de la barrera hematoencefálica (BHE) y necrosis isquémica.34,35
Respuesta inflamatoria y disfunción endotelial
La circulación elevada de ALA y PBG conlleva a daño tisular, debido a la liberación de patrones moleculares de señalización inflamatoria.32,36 Esto activa la secreción de interleucinas (IL-1b, IL-2, IL-6, IL-17), INF-y, TNF, complemento, factor de crecimiento endotelial vascular, que conducen a la expresión de especies reactivas de oxígeno (ERO) y el daño generalizado.32,35 Las ERO, en conjunto con el factor de transcripción nuclear, factor kappa B (NF-κB), promueve más inflamación, aumenta la permeabilidad endotelial, que lleva a edema cerebral intersticial y el riesgo de SEPR.37,38 Además, esta reacción inflamatoria afecta el transporte axonal y favorece la degeneración axonal.3,30
Neurotoxicidad por productos de porfirinas en el SNC
La BHE es impermeable en un 95 % a estos precursores de ALA y PBG.32 El movimiento de ALA a través de la BHE se debe al sistema de transporte por difusión pasiva, regulado por el transportador de péptidos 2 (PEPT2) tipo 1 y 2.32,39 El PEPT2 cataliza el transporte al acoplar su translocación con un gradiente transmembrana tipo 1 (alta afinidad) - tipo 2 (baja afinidad), mantiene los niveles de ALA en el fluido cerebral espinal bajo, en comparación con el plasma y ejerce un efecto neuroprotector.32,40 Los PEPT2 tipo 2 por su baja afinidad tienen un flujo cerebral bajo con un menor gradiente de difusión de ALA hacia el plasma, que lleva a una neurotoxicidad más significativa.14,30
Se ha descrito la interacción entre ALA, el ácido gamma-amino butírico (GABA) y L-glutamato.14 Esto se debe a la similitud entre sus estructuras, dado que el ALA al interactuar con los receptores GABA y L-glutamato, antagoniza los efectos inhibitorios en el SNC y desregula el proceso de trasmisión nerviosa, lo cual desencadena los episodios de convulsiones.3,34
Disfunción bioenergética mitocondrial y lesión neuromuscular
Las mitocondrias son orgánulos de doble membrana que proporcionan energía para las células, señalización del calcio, apoptosis celular, regulación del ERO, además modulan la plasticidad sináptica.41,42,43 El hemo es un cofactor importante para los citocromo c, en los complejos II, III, IV del transporte mitocondrial, por lo que su disminución genera disfunción mitocondrial.33,36,44 La acumulación de ALA reduce las actividades enzimáticas de los complejos de la cadena respiratoria, que disminuyen la producción de adenosina trifosfato (ATP), aumentan la neurotoxicidad celular, el desarrollo de edema cerebral y daño de la placa neuromuscular.3,32 La falla bioenergética, además puede afectar procesos dependientes de la energía, como la función PEPT2 y el Na+ en células gliales, que modifica la homeostasis del Ca++, intensifica la producción de ROS e induce apoptosis de células nerviosas.32,42
Lesión tubular renal por productos de porfirina
Los niveles elevados de ALA y PBG, aumentan la tasa de filtración de estos productos, que son reabsorbidos en los túbulos renales proximales y llevan al daño.39 La reabsorción se debe a la acción de los PEPT1 tipo 2 en los segmentos S2-S3 de los túbulos renales proximales hacia el interior de la célula, que incrementa la síntesis de uroporfirinógeno, ERO y secreción de citocinas proinflamatorias como la IL-6.25 Esto conduce a un efecto citotóxico que promueve cambios fenotípicos epiteliales, apoptóticos, regeneración y cicatrización.39 Estas lesiones se reflejan en nefropatía tubulointersticial primitiva aguda, que favorece la isquemia y lesión renal durante los ataques súbitos.45)
Manifestaciones clínicas
El reconocimiento de una crisis aguda sin un diagnóstico conocido de porfiria es difícil, debido a síntomas y signos inespecíficos.7,46 En la fase aguda, las manifestaciones son neuroviscerales, como taquicardia (65-85 %), hipertensión arterial (35-55 %), dolor abdominal (85-95 %), emesis (50 %), constipación (50 %), neuropatía periférica (42-68 %) y alteraciones neuropsíquicas asociadas - trastornos en la conducta.2,20,47 En los ataques graves, progresa con paresia localizada de los miembros superiores e inferiores (40-65 %), parálisis bulbar, polineuritis con lesión de los nervios craneales, convulsiones y parálisis respiratoria (9-20 %), que compromete la vida de los pacientes.7,20,28) Las lesiones en la piel se manifiestan como erupciones ampollosas cutáneas fotosensibles crónicas, propias de CPH y la PV.15
Porfiria aguda intermitente
Las personas que cursan con PAI se clasifican como sintomáticas (15-20 %) y asintomáticas (80-85 %).37 Aproximadamente 5-8 % de los pacientes sintomáticos, tienen ataques recurrentes (≥ 4 ataques por año).7,10 Los episodios agudos inician con dolor abdominal agudo grave (85-95 %), emesis (50 %), estreñimiento (55 %) y síntomas neurológicos.8,28,35 Las alteraciones neuropsíquicas incluyen ilusiones, alucinaciones, alteraciones del pensamiento (24-70 %) y episodios delirantes (40 %).28,47 Las convulsiones ocurren entre el 10-20 % de los pacientes con PAI exacerbadas, con hiponatremia moderada.3,48 Algunos pacientes presentan polineuropatía; se considera un síntoma de presentación confusa y difícil distinguir de la polineuropatía secundaria al síndrome de Guillain-Barré.19,40 Sin embargo, la PAI afecta las extremidades superiores, en lugar de la parálisis ascendente característica del Guillain-Barré.40 La neuropatía periférica puede progresar a parálisis bulbar y la parálisis muscular respiratoria, lo que lleva a la necesidad de soporte de ventilación mecánica invasiva, que aumenta el índice de mortalidad.19,42
Porfiria variegata y coproporfiria hepática
En la CPH las crisis agudas son más leves que las otras porfirias hepáticas.20 En fase aguda aparecen síntomas neuroviscerales, que debutan después de la pubertad.28 La crisis aguda inicia con síntomas abdominales emesis, náuseas, diarrea, dolor epigástrico (89 %), neurológicos (33 %) y cardíacos (25 %).28
En la PV se encuentran síntomas neuroviscerales, acompañados de manifestaciones cutáneas muy parecidas a la porfiria cutánea tardía.20 Los síntomas neuroviscerales debutan con dolor abdominal, estreñimiento, taquicardia, hipertensión, ansiedad, agitación, convulsiones y debilidad neuromuscular que puede progresar a cuadriparesia y parálisis respiratoria.22 Las manifestaciones cutáneas, son lesiones ampollas, vesículas subepidérmicas, ampollas y erosiones que forman costras y cicatrizan lentamente.28 Los cambios en la piel se limitan a zonas expuestas al sol y están clínicamente presentes de las otras porfirias cutáneas.20
Porfiria por deficiencia de ácido delta aminolevulínico deshidratasa
Es un trastorno de baja prevalencia, con inicio en la infancia e implicaciones neurológicas graves.33 Los pacientes inician con síntomas de emesis, taquicardia, náuseas y dolor abdominal, pero a excepción de la CPH y la PV, no presentan lesiones cutáneas.22
Diagnóstico
Las porfirias agudas pueden prevalecer sin diagnosticar hasta 10-15 años después del inicio de los síntomas debido a su inespecificidad, que imitan otros trastornos comunes.5 La evaluación inicial incluye una historia clínica detallada, examen físico, con evaluación neurológica.31 Se deben solicitar pruebas de laboratorio bioquímicas, genéticas para identificar el tipo específico de porfiria.7
Pruebas bioquímicas
Se solicita un conteo sanguíneo completo, para analizar células sanguíneas, electrolitos, índices renales, función hepática y niveles de hierro.31 La hiponatremia es una manifestación metabólica (25-60 %) secundaria a la pérdida excesiva de sodio, debido a síntomas gastrointestinales y diuresis osmótica después de infusión de glucosa sin reemplazo de electrolitos.6 Las aminotransferasas séricas están elevadas (13 %) en los pacientes durante la crisis aguda.31 Ante la sospecha de porfiria hepática, la prueba de orina es ideal para el diagnóstico inicial.49 Los niveles de ALA y PBG son eliminados hasta 10 veces por encima del límite normal, el cual es observable mediante la prueba cualitativa de Watson-Schwartz o Hoesch.49,50 Consiste en la formación del complejo urobilinógeno - Ehrlich después de añadir cloroformo, lo que genera una tonalidad roja a violeta, pero tiene baja sensibilidad.8,19) Sin embargo, niveles de PBG urinario 1 a 2 veces por encima de 4 mg/L no es específica para el diagnóstico de porfiria aguda hepática.5 En las crisis por CPH - PV, una muestra de 5-10 g de heces fecales, pueden ser métodos electivos durante la crisis aguda. Se puede hallar coproporfirina III y protoporfirina IX en heces, por lo se pueden solicitar ante la sospecha de porfirias hepáticas.25
Pruebas genéticas
Las pruebas genéticas son el gold standard para el diagnóstico específico de las porfirias agudas.4) Una vez que se confirma bioquímicamente el diagnóstico, se requiere la secuenciación de genes, para identificar el tipo de mutación y el tamizaje familiar para identificar los portadores.31 Esta detección de las mutaciones se lleva a cabo mediante la amplificación por reacción en cadena de la polimerasa (PCR) de los exones ALAS1, ALAS2, ALAD, HMBS, CPOX y PPOX.3
Tratamiento
La crisis aguda constituye una urgencia médica y requiere tratamiento hospitalario con enfoque integral para disminuir la actividad ALAS1, los síntomas neuroviscerales y anormalidad electrolítica.3) Se puede requerir la admisión en la unidad de cuidados críticos para el tratamiento de soporte respiratorio mecánico invasivo, convulsiones y la infección precipitante.51
Tratamiento inicial
La administración de dextrosa es el paso inicial.5 La infusión venosa de altas dosis de glucosa, reprime el ARNm de la ALA-S1 a través del coactivador del receptor activado, por el proliferador de peroxisomas 1α.37 Si el paciente tolera la vía oral; iniciar dieta alta en carbohidratos durante 48 horas antes de comenzar un tratamiento específico.5 En la actualidad, la hemina intravenosa es la terapia más efectiva para los episodios agudos.52 La dosis de la infusión de hemina (3-4 mg/kg i.v. al día por 4 días en 30 min) permite la corrección del déficit de hemo.16 Sin embargo, las preparaciones de hemina no están disponibles en la mayoría de centros hospitalarios en todo el mundo.52
Las infusiones profilácticas de hemina individualizadas, semanales y mensuales, son beneficiosas, en pacientes que sufren ataques recurrentes, pero las pautas específicas para la frecuencia óptima de administración y la interrupción de la terapia profiláctica, no están definidas.11,53 Pueden dar lugar a síntomas crónicos, particularmente dolor neuropático crónico y neuropatía progresiva, que deterioran notablemente la calidad de vida, que hace necesario otras terapias alternativas.11
El tratamiento del dolor debe abordarse de inmediato, debido a que el estrés por dolor contribuye a reacciones neuroendocrinas que activan ALAS1 y exacerban los síntomas.31 Los opiáceos parenterales son la mejor opción (morfina, diamorfina y fentanilo) y la gabapentina.25 El uso de betabloqueantes (propanolol), es capaz de frenar la hiperactividad simpática. Las náuseas y los vómitos son controlables con proclorperazina, promazina y ondansetrón; disminuye de 72 a 96 horas.5
El tratamiento de los episodios de convulsiones es difícil en pacientes con PAI.3) Muchos de los medicamentos antiepilépticos convencionales (fenitoína, carbamazepina, primidona y ácido valproico) son potencialmente porfirinógenos, lo que complica la evolución del paciente.48 Actualmente las benzodiacepinas, gabapentina y el levetiracetam, son fármacos seguros utilizados durante la crisis aguda.54 Para el estatus epiléptico no controlado, se ha informado que la infusión de propofol es segura.3
Trasplante ortotópico hepático (TOH)
En la actualidad es el único tratamiento curativo disponible, con restablecimiento de la actividad enzimática dentro de las 24-72 horas.46 Aunque tiene alta morbilidad y mortalidad, está indicado primordialmente en pacientes que debutan con crisis aguda posterior a PAI.20 En los pacientes que se trasplantaron con éxito, los niveles de ALA y PBG en la orina se encontraban reducidos, por tal razón este tratamiento disminuye el riesgo de presentar ataques agudos, hasta por 10 años aproximadamente.11 No es de primera opción, debido a las complicaciones asociadas y la dependencia de terapia inmunosupresora de por vida.55)
Terapia génica (perspectivas actuales)
Debido a la alta incidencia de fracaso de las infusiones de hemina y las complicaciones generadas por TOH, muchos investigadores han optado por la terapia génica como opción alternativa no invasiva.46,56 La interferencia de ARN, consiste en un proceso natural que regula la expresión de un gen específico en una determinada secuencia, lo cual suprime la proteína que causa la enfermedad.10,57 Esta capacidad de suprimir selectivamente genes, se ha convertido en un enfoque terapéutico prometedor y validado para el tratamiento de los pacientes con porfirias hepáticas.58 En un una investigación realizada por Sardh y otros.10 de fase 1 en pacientes con PAI, el uso de un inhibidor de ARN (givosiran) redujo los niveles de ALA, PBG y ARNm de ALAS1, a niveles casi normales, con disminución del uso de hemina. Después de la inyección subcutánea, el givosiran es endocitado por los hepatocitos que llevan el receptor de asialoglicoproteína de unión a N-acetil-galactosamina.25 En un ensayo clínico en fase 1 de D’Avola D y otros,56 utilizando terapia dirigida al vector AAV (rAAV2/5-PBGD) en 8 pacientes, el efecto del tratamiento sobre los resultados clínicos fue muy variable, se encontró una mejoría en 2 pacientes, en el resto el efecto beneficioso fue bizarro.
Pronóstico
Debido a la baja incidencia y la duración de la detección del diagnóstico, existe poca evidencia investigativa y datos limitados acerca del pronóstico.59 Durante las crisis agudas, la administración de fármacos para contrarrestar algunos síntomas pueden agravar la condición clínica y llevar al desenlace fatal.8) La mortalidad ha disminuido en las últimas décadas del 5-20 % gracias a nuevas herramientas de diagnóstico y tratamiento.5 Si existe fallo en la terapia con hemina e inhibidores de ALAS1, se recomienda el trasplante hepático.5 El monitoreo continuo ambulatorio, debe ser individualizado y esencial para prevenir ataques agudos, hospitalización y complicaciones a largo plazo.31 Los pacientes que se programan para procedimientos quirúrgicos, deben informar al cirujano de su diagnóstico, para proporcionar el tiempo adecuado para la evaluación, planificación de los distintos insumos y anestésicos que se utilizarán durante la cirugía.31
La presentación clínica de las porfirias hepáticas es muy variable, debido a la inespecificidad de sus síntomas. Sin embargo, las pruebas bioquímicas y genéticas están cada vez más al alcance del personal médico para la confirmación diagnóstica, reducen el tiempo entre la sospecha y el tratamiento. Debido a la dependencia de la hemina de por vida, se ha optado por nuevas alternativas terapéuticas, como la supresión genética y el trasplante hepático. El pronóstico es favorable cuando se realiza el diagnóstico a tiempo.

