










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El penfigoide ampolloso (PA) es una dermatosis vesicular ampollosa, autoinmunitaria, subepidérmica, que afecta principalmente a adultos mayores de 70 años. Se caracteriza por la formación de ampollas subepidérmicas tensas y pruriginosas en tronco y extremidades, predominantemente.1 Se estima una incidencia que oscila entre 2,5 a 42,8 casos por cada millón de habitantes al año, en el mundo.2,3 La explicación fisiopatológica de la enfermedad, es que se debe a la formación de autoanticuerpos que reconocen autoantígenos en la zona de la membrana basal.2
Múltiples enfermedades pueden simular la formación de ampollas pruriginosas, entre los cuales se encuentran la urticaria, prurigo, impétigo, eritema multiforme, necrólisis epidérmica tóxica; son ampliamente reconocidas como diagnósticos diferenciales del PA.1,3 El tratamiento inmunomodulador con corticoides por vía enteral o parenteral siguen siendo el tratamiento de primera línea, además del control de enfermedades concomitantes o comorbilidades.1,4
En la actualidad es limitada la literatura médica sobre las principales manifestaciones clínicas, diagnóstico y tratamiento del PA,4 por lo cual es necesario promover la descripción, análisis y reportes de casos clínicos, para ampliar el conocimiento científico en el personal médico.
El objetivo de este trabajo es presentar el caso clínico de un paciente con diagnóstico de penfigoide ampolloso de origen idiopático.
CASO CLÍNICO
Paciente masculino de 81 años con antecedentes de tabaquismo, quien acude al servicio de urgencias por cuadro clínico de 2 meses de evolución, consistente en la aparición de lesiones ampollosas, dolorosas, generalizadas y síndrome de pérdida involuntaria de peso.
Al examen físico se evidencian ampollas tensas, dolorosas a la palpación, de contenido serohemático, de tamaño variado, bordes regulares y base eritematosa, asociadas a erosiones y costras hemorrágicas que comprometían toda la superficie corporal (Fig. 1).
El hemograma al ingreso muestra elevación de leucocitos, neutrófilos y eosinófilos, gases arteriales con alcalosis respiratoria, con un trastorno moderado de la oxigenación. Por lo anterior se considera como primera posibilidad diagnóstica PA frente a un síndrome hipereosinofílico, por lo cual se realizan exámenes de laboratorio de control y una biopsia de piel para confirmar el diagnóstico.

Un hemograma a las 72 horas después del ingreso revela persistencia en los altos niveles de leucocitos, neutrófilos y eosinófilos; los hemocultivos, al cuarto día de incubación informa presencia de Streptococcus intermedius. Ante la posibilidad diagnóstica de un PA, con actividad importante y sobreinfección bacteriana se inicia antibioticoterapia empírica, con clindamicina, 600 mg cada 6 h.
Una electroforesis de proteína presenta un pico en la zona gamma, la cual se relaciona con el proceso infeccioso. Las imágenes diagnósticas de tórax, abdomen y pelvis no tenían hallazgos sugestivos de procesos neoplásicos. Ante el empeoramiento clínico, dado por lesiones en la piel, con afectación extensa de la superficie corporal, mayor del 30 % (Fig. 2), se traslada a la unidad de cuidados intermedios, por alto riesgo de insuficiencia cutánea e inician tratamiento inmunomodulador con corticoide por vía enteral.
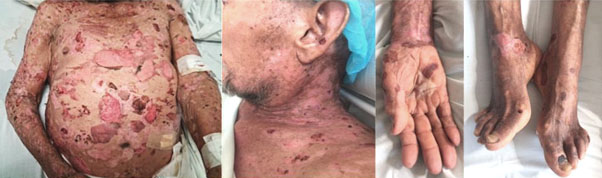
El resultado de la biopsia de piel de la axila izquierda reporta dermatitis perivascular, con eosinofilia tisular y ampolla subepitelial focal, inmunoglobulina A positivo y complemento positivo. El paciente culmina tratamiento antibiótico durante 14 días, con control de la respuesta inflamatoria y cultivos negativos. El control de las lesiones en la piel permitió realizar el egreso hospitalario con tratamiento corticosteroide por vía oral y seguimiento ambulatorio por medicina interna y dermatología, ante el diagnóstico de PA de origen idiopático.
COMENTARIOS
La lesión crónica de la unión dermoepidérmica por autoanticuerpos está mediada por elastasas neutrofílicas, metaloproteinasas de la matriz, linfocitos activados y células presentadoras de antígenos.2,3 Los eosinófilos juegan un rol fundamental en el PA, dado por la infiltración de eosinófilos y de granulación, precedido por la formación de ampollas y daño tisular evidentes en las biopsias de piel;5 además, una correlación directamente proporcional entre el número de eosinófilos periféricos y los niveles de anticuerpos inmunoglobulinas E, lo cual es característico en la enfermedad.5,6
En los exámenes de laboratorio del paciente, al inicio se mostró un aumento marcado en los eosinófilos, junto a dermatitis perivascular con eosinofilia tisular y ampolla subepitelial focal, sin detección de inmunoglobulina E en la histopatología de la lesión. Se enfocó un diagnóstico inicial de síndrome hipereosinofílico o neoplasia hematolinfoide, sin embargo, el hemocultivo positivo para infección bacteriana y empeoramiento de las lesiones ampollas subepidérmicas, inclinó el diagnóstico diferencial hacia una dermatosis penfigoide tipo PA.7
En la literatura se describen diversas variantes clínicas, entre ellas el penfigoide no ampolloso, PA clásico, penfigoide de membranas mucosas y epidermólisis ampollosa adquirida.2 El penfigoide no ampolloso se manifiesta con lesiones pruriginosas en forma de parches eccematosos, similares a la dermatitis atópica y urticaria. La fase ampollosa se manifiesta con lesiones vesiculoampollosas tensas, que aparecen más comúnmente sobre una base eritematosa y predominan en las zonas de flexión; la afectación de las mucosas se observa en menos del 30 % de los pacientes; la afectación mucosa de la cavidad oral se presenta en hasta en el 85 % de los casos.4,8 La epidermólisis ampollosa adquirida se diferencia del PA ampolloso clásico, debido a que tiene afectación en áreas susceptibles al trauma, como codos y rodillas, además de afectación en la orofaringe.1,8 Las lesiones vesiculoampollosas tensas se presentaron en áreas extensoras típicas de la epidermólisis ampollosa adquirida, sin embargo, en el paciente que se presentó no se evidenciaron lesiones en áreas mucosas ni en la orofaringe, relacionadas con una variante de penfigoide ampolloso clásico.
El reconocimiento a través de las manifestaciones clínicas es indispensable en el enfoque diagnóstico y terapéutico inicial, favorece en la morbimortalidad y pronóstico de la enfermedad.2,4 La histopatología de la piel perilesional puede demostrar 2 patrones histopatológicos: un tipo clásico no inflamatorio o un tipo inflamatorio con neutrófilos mixtos, linfocitos y eosinófilos y la detección de anticuerpos séricos contra BP180 o BP230 mediante ensayo inmunoabsorbente ligado a enzimas.9 La inmunofluorescencia directa presenta un depósito lineal de anticuerpos de inmunoglobulinas (IgM, IgA, IgG) y proteínas del complemento (C3) en la zona de la membrana basal.2
El objetivo del tratamiento es el control de las manifestaciones clínicas, consecuencia de una respuesta inflamatoria descontrolada. El tratamiento de primera línea para la PA incluye corticosteroides tópicos de alta potencia. En pacientes con lesiones generalizadas se pueden considerar los corticosteroides sistémicos por vía oral o parenterales.4 Las terapias de segunda línea son los inmunosupresores, que incluyen azatioprina, micofenolato de mofetilo, metotrexato, ciclofosfamida, entre otros.1,4 Por último, la elevación en los niveles de anticuerpos inmunoglobulina E se presenta en más del 70 % de los pacientes con PA, por lo cual pueden utilizar anticuerpos monoclonales humanizados, como el omalizumab o dupilumab en el tratamiento inmunomodulador de la enfermedad.10
El penfigoide ampolloso es poco frecuente, con una elevada tasa de mortalidad si se realiza un diagnóstico y tratamiento tardío. Reconocer las principales manifestaciones y variantes clínicas de esta enfermedad permite un oportuno enfoque diagnóstico y terapéutico, este último basado en el control de la respuesta inflamatoria contra la piel y otros órganos.

