










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La leucoencefalopatía multifocal progresiva (LMP) es una enfermedad desmielinizante del sistema nervioso central de etiología viral.1 La enfermedad fue descrita en 1958 por Astrom, Mancall y Richardson en dos pacientes con leucemia y otro con enfermedad de Hodking. En 1971 se identifica el virus como perteneciente a la familia papovaviridae, género poliomavirus y se le asigna la sigla JC (iniciales del paciente John Cunningham en quien fue aislado el virus por primera vez).2
JC es un virus distribuido en todo el mundo y afecta más del 50% de la población mundial. La infección por el virus JC, se produce en la infancia; es en general asintomática y el virus queda latente en parénquima renal, médula ósea y tejido linfoide. En condiciones de inmunodepresión, el virus se replica y adquiere potencial neuropatógeno.3
El hallazgo neuropatológico fundamental de la LMP es la desemielinización; esta es consecuencia de la infección directa de los oligodendrocitos por el virus JC. La replicación viral activa y la formación de la cápside en oligodendrocitos infectados es seguida de citólisis y liberación de viriones a células vecinas, lo cual conlleva a la destrucción de estas células y su función principal que es la producción de mielina.4
Desde el año 1983, y en relación con la infección por el virus de inmunodeficiencia humana (VIH), aumentaron los pacientes con LMP constituyendo el 4% de los casos neurológicos asociados a VIH, sobre todo en pacientes severamente inmunosuprimidos, con conteo de linfocitos T CD4 inferiores a 200 células/mm3.5) Sin embargo, en las últimas décadas se ha descrito una variante de LMP asociada a medicamentos inmunomoduladores y también en pacientes con neoplasias, enfermedades inflamatorias y autoinmunes como artritis reumatoide y lupus eritematoso sistémico, con una mortalidad del 75 %. En estos casos, los pacientes presentan manifestaciones neurológicas que resultan complicadas de diferenciar entre las propias producidas por las enfermedades autoinmunes o por una posible LMP, esto conlleva al retraso en el diagnóstico y por lo general, a la muerte del paciente.6
Los síntomas clínicos de LMP, dependen del lugar dañado en el sistema nervioso central. Las regiones frontales y parieto-occipitales son las que más se afectan y la lesión en fosa posterior, es solo descrita en el 34 % de los casos.1
Se reporta un paciente con diagnóstico de infección por VIH/sida asociada a LMP cerebelosa; con el objetivo de orientar sobre la posibilidad diagnóstica de LMP ante manifestaciones neurológicas de pacientes inmunodeprimidos.
Caso clínico
Antecedentes y enfermedad actual
Se trata de un paciente masculino, de 25 años de edad, blanco, de procedencia urbana, con antecedentes de salud aparente. Según refiere, 1 año previo a su ingreso, le apareció una lesión blanquecina en la lengua y cerca de las encías, con aspecto grumoso y no buscó ayuda médica. El paciente comienza con lenguaje disártrico y dificultad para caminar tambaleándose hacia los laterales, refiriendo que en ocasiones chocaba con las paredes, puertas, u otro obstáculo, que se encontrara en su camino. Presenta, además, dificultades para percibir los estímulos de dolor, calor, vibración, incluso se quejaba con mucha frecuencia de calambres musculares; otras veces quería alcanzar un objeto y le era difícil el agarre. A todo esto, se suma, inapetencia, astenia marcada, pérdida de peso de 14 kg y febrícola esporádica en diferentes horarios, no constatada por termómetro. Este cuadro tuvo una instauración progresiva y lenta, durante de tres meses, posterior a los cuales nota empeoramiento y decide solicitar ayuda médica.
Exploración física positiva
Sistema nervioso central: Consciente, orientado en tiempo, espacio y persona, no signos meníngeos, con defecto motor asociado (dismetría, marcha atáxica). Maniobra Índice-nariz, e Índice-índice positivas. Disdiadocinesia: Temblor mixto generalizado, lenguaje escandido e hiperreflexia.
Con estos elementos se ingresa en centro hospitalario para investigación clínica, diagnóstica y tratamiento oportuno.
Pruebas complementarias positivas
Eritrosedimentación: 70 mm/h (Valor de Referencia (VR) hombre hasta 20 mm/h).
Carga viral de VIH plasmática: 161 000 copias/mL. Log: 5,21.
Conteo de linfocitos T CD4: 2 %-9 células/mm3. (VR 500-1000 células/mm3).
Estudio del líquido cefalorraquídeo (LCR): Reacción en cadena de polimerasa (PCR) positivo para virus JC.
Tomografía axial computarizada (TC) de cráneo contrastada no encontrándose lesión tumoral, no lesión isquémica aguda, ni lesión hipercaptante.
Imagen por resonancia magnética (IRM) de cráneo (Fig.): se realizaron secuencias T1 y FLAIR axial (significado en inglés: Fluid-Alternated inversión recovery) y T2 sagital con cortes de 6 mm donde se observa marcado engrosamiento en seno maxilar izquierdo y en celdas etmoidales izquierdas. Presenta aumento de la amplitud de las folias cerebelosas en relación con atrofia cerebelosa provocando leve dilatación compensadora del 4to ventrículo.
En FLAIR se observan lesiones hiperintensas en el hemisferio cerebeloso derecho y pedúnculo cerebeloso medio izquierdo, las que se consideran lesiones sugestivas de Leucoencefalopatía multifocal progresiva (LMP) en tallo encefálico y cerebelo (Fig.).
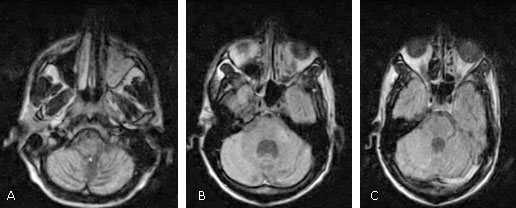 A: muestra signos de atrofia cerebelosa y marcado engrosamiento mucoso en seno maxilar izquierdo. B: lesiones hiperintensas en hemisferio cerebeloso derecho y en pedúnculo cerebeloso medio izquierdo de aspecto desmielinizante. Dilatación compensadora del 4to. ventrículo. C: lesión hiperintensa en hemisferio cerebeloso derecho. Engrosamiento mucoso en celdas etmoidales izquierdas.
A: muestra signos de atrofia cerebelosa y marcado engrosamiento mucoso en seno maxilar izquierdo. B: lesiones hiperintensas en hemisferio cerebeloso derecho y en pedúnculo cerebeloso medio izquierdo de aspecto desmielinizante. Dilatación compensadora del 4to. ventrículo. C: lesión hiperintensa en hemisferio cerebeloso derecho. Engrosamiento mucoso en celdas etmoidales izquierdas.Fig Imagen por resonancia magnética de cráneo, secuencia FLAIR en plano axial A-C.
Evolución
Teniendo en cuenta las características clínicas, neuroimagen (IRM), la presencia de virus JC en LCR confirmado por PCR y la infección demostrada de VIH; se realiza diagnóstico de LMP, como forma de debut clínico de VIH, se comienza con terapia antiretroviral, logrando controlar carga viral de VIH y recuperación inmunológica en los tres meses posteriores al diagnóstico, en los cuales, no hubo mayor progresión de la LMP.
Discusión
Leucoencefalopatía multifocal progresiva (LMP) es una enfermedad descrita en pacientes inmunodeprimidos. Tradicionalmente asociada a VIH/sida, sin embargo, en las últimas décadas se ha descrito el término de LMP asociada a medicamentos, debido al incremento del uso de medicamentos inmunomoduladores en la esclerosis múltiple, enfermedades reumáticas e inflamatorias de larga evolución como sarcoidosis, lupus eritematoso sistémico y otras.7
El caso presentado constituye un ejemplo de la forma de presentación de LMP, como enfermedad oportunista que permite el diagnóstico de una enfermedad inmunosupresora subyacente. De ahí, la importancia de tener en cuenta LMP, no solo en el paciente con antecedentes conocidos de inmunosupresión, sino, en todo aquel paciente con manifestaciones neurológicas, pues se reporta en la literatura, que LMP constituye una entidad inicial en el diagnóstico de enfermedades inmunosupresoras como VIH, artritis reumatoide, lupus eritemtoso sistémico, sarcoidosis y otras dentro de grupo de las enfermedades inflamatorias y autoinmunes.8
Las manifestaciones clínicas de LMP, son fundamentalmente neurológicas. El diagnóstico se plantea ante un paciente con cuadro de deterioro cognitivo o déficit focales (hemiparesia, alteraciones campimétricas, alteraciones de la coordinación y del equilibrio, etcétera), o ambos, de curso progresivo, en cuestión de semanas o pocos meses.9
El espectro clínico de manifestaciones neurológicas es amplio y dependen de la localización de la desmielinización, siendo más frecuentes en región parieto -occipital. En el presente caso, las lesiones tuvieron mayor localización en fosa posterior, con evidencias de daño en cerebelo y tallo encefálico, lo que se corresponde con las manifestaciones clínicas descritas en el paciente. Scholten reporta que solo 10 % de los pacientes presenta localización cerebelosa o en tronco encefálico.1
La tomografía computarizada (TC) encefálica puede ser normal al inicio de la enfermedad, o mostrar lesiones hipodensas de la sustancia blanca, a menudo confluentes, más frecuentemente ubicadas en las regiones frontales y parietooccipitales que no se realzan con el contraste, no producen efecto de masa y respetan la sustancia gris cortical, aunque pueden afectar la sustancia gris gangliobasal.10
En el paciente que se ha presentado, tampoco hubo evidencias de lesiones en la TC, por tanto, es importante destacar, que estos pacientes pueden tener lesiones orgánicas con TC de cráneo negativas, debido a que la TC posee mayor sensibilidad para lesiones coalescentes de gran tamaño; las lesiones de pequeño tamaño, como las producidas en LMP, no producen efecto de masa y por tanto es difícil su observación mediante TC.
El estudio de mayor sensibilidad para mostrar las lesiones desmilinizantes es la resonancia magnética de cráneo. Permite observar mejor las lesiones de LMP y su extensión, las cuales aparecen de baja intensidad en T1 e hiperintensas en T2 y FLAIR, localizadas en sustancia blanca subcortical que se extienden a sustancia blanca profunda, con predilección por afectar las fibras en U subcorticales. No producen efecto de masa y no se refuerzan con el gadolinio.11
El consenso sobre criterios diagnósticos de enfermedades neuroinfecciosas publicado por Berger et al, define la triada necesaria para diagnóstico de LMP: partiendo de la condición previa de inmunosupresión hay que tener en cuenta características clínicas, neuroimagen sugerente de enfermedad desmielinizante y confirmación de presencia del virus JC en LCR, mediante la técnica de reacción en cadena de la polimerasa.12 Dicha técnica, para el virus JC, tiene una sensibilidad de 65 %, y una especificidad de 92 %.2
El caso presentado, evidencia una forma de presentación atípica de LMP, con afectación de fosa posterior (cerebelo y de tallo encefálico). Para el diagnóstico, resultaron positivos los tres elementos que constituyen la triada diagnóstica: las características clínicas con neuroimagen (RM) sugerente y un PCR para virus JC, positivo en el LCR.
A modo de conclusión, LMP debe incluirse en el diagnóstico diferencial en todo paciente con manifestaciones neurológicas de afectación en fosa posterior y estudiar causas de inmunosupresión subyacente. La localización cerebelosa es infrecuente, pero puede observarse en aproximadamente el 10 % de los casos. La triada de características clínicas, neuroimagen sugerente y aislamiento del virus en el LCR, conforman los criterios diagnósticos.

