










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter v.17 n.3 Ciudad de la Habana sep.-dic. 2001
Instituto de Hematología e Inmunología
Agammaglobulinemia ligada al X o de Bruton. Aspectos clínicos, moleculares y terapéuticos
Dra. Vianed Marsán Suárez
Resumen
La agammaglobulinemia ligada al X o de Bruton constituye el prototipo de deficiencia primaria de célula B. Los niños varones afectados presentan infecciones recurrentes y manifestaciones autoinmunes a partir de los 6 meses de edad. La utilización de modernas técnicas de biología molecular ha permitido la identificación del gen responsable de la enfermedad en el locus Xq22. La naturaleza genética de la misma ha posibilitado además, la detección de madres portadoras y la realización de un diagnóstico prenatal. Actualmente se continúa en la profundización de los aspectos moleculares, con el objetivo de manipular el material genético de los pacientes con fines terapéuticos, lo que resultará en una cura definitiva de la enfermedad.
DeCS: AGAMMAGLOBULINEMIA/diagnóstico; CROMOSOMA X/inmunología; LINFOCITOS B; PROTEINO-TIROSINA QUINASA/deficiencia.
Las inmunodeficiencias primarias (IDP) son desórdenes congénitos que afectan la función del sistema inmunológico, lo que provoca una respuesta inmune inadecuada a microorganismos patógenos, antígenos propios y células tumorales, que se manifiesta clínicamente en un incremento de la susceptibilidad a las infecciones, enfermedades alérgicas, neoplásicas y autoinmunes.1,2
Las anomalías congénitas en el desarrollo y la función de los linfocitos B, dan lugar a una producción defectuosa de anticuerpos (Acs). Los pacientes con estos desórdenes presentan infecciones recurrentes por bacterias piógenas como neumococo, estafilococo, estreptococo y Haemophilus. Además, son susceptibles a ciertas infecciones virales, como la poliomielitis y a algunos parásitos intestinales como la Giardia lamblia, lo que demuestra el papel importante de los Acs en la adherencia, opsonización y fagocitosis de estos organismos.3,4
La agammaglobulinemia ligada al X (ALX) fue la primera IDP identificada en 1952 por Bruton, por lo que también es llamada agammaglobulinemia de Bruton, y constituye el prototipo de déficit de células B. Es una enfermedad ligada al cromosoma X, de forma tal que las mujeres que presentan el gen defectuoso en uno de sus cromosomas X son normales, pero cuando lo transmiten a sus hijos varones, estos manifiestan la enfermedad.4,5
Diagnóstico
Los niños varones con ALX comienzan a presentar infecciones bacterianas recurrentes a partir de los 6 meses de edad, tiempo en que los Acs maternos adquiridos pasivamente comienzan a ser catabolizados.6
Estos pacientes presentan infecciones causadas fundamentalmente por bacterias grampositivas que colonizan diferentes órganos, principalmente oído medio, bronquios, pulmones, conjuntivas, piel y meninges; pueden además ser infectados en menor frecuencia por enterovirus y virus de la hepatitis. El 20 % de los casos presenta artritis séptica que puede estar asociada o no con un síndrome de malabsorción intestinal. Se ha descrito en algunos pacientes la aparición de manifestaciones autoinmunes, entre las que se encuentran la colitis ulcerativa, una artritis similar a la reumatoide juvenil y anemia hemolítica autoinmune.7-9
En el examen físico de estos niños es característico encontrar la ausencia de tonsilas y adenopatías, así como una disminución del peso y la talla para su edad.4
El diagnóstico clínico debe ser corroborado por la demostración de la ausencia o la marcada disminución de las 5 clases de inmunoglobulinas (Igs) en sangre periférica, mediante una electroforesis de proteína, cuantificación de Igs e inmunoelectroforesis. El nivel serológico de la IgG total usualmente se encuentra por debajo de 250 mg/dL, el resto de los isotipos M, A, D y E, se encuentran extremadamente bajos o no son detectados. La respuesta de Acs específicos tras estimulación con diferentes antígenos está marcadamente deprimida.4,7,10
Este diagnóstico se hace difícil antes de los 6 meses de edad, debido a la presencia de Acs maternos; en este caso, una biopsia intestinal que muestre la ausencia de células plasmáticas en la lámina propia del intestino, pudiera ser indicada para confirmar el diagnóstico; sin embargo, por lo invasivo del proceder, se recomienda repetir los estudios en el segundo semestre de vida.4,11
Estos pacientes presentan una respuesta celular conservada, la cuantificación de subpoblaciones linfocitarias T, la prueba de hipersensibilidad retardada, la linfoproliferación frente a la fitohemaglutinina y el cultivo mixto de linfocitos muestran valores normales; de igual forma, la actividad de las células asesinas naturales es normal.11
Diagnóstico diferencial
Para el diagnóstico definitivo de una ALX es necesario descartar otras enfermedades que presentan comportamientos clínicos y serológicos bastante similares, entre las que se encuentran: la hipogammaglobulinemia fisiológica (HF) o transitoria de la infancia (HTI), la malabsorción intestinal severa (MIS), la artritis juvenil (AJ) y la fibrosis quística (FQ).
Durante los 3 a 6 meses de vida, el niño presenta una HF como consecuencia de la disminución paulatina de la IgG materna adquirida a través de la placenta y el inicio progresivo de la producción propia de Acs; en el caso de que esta última se retrase, aparece la denominada HTI, que puede persistir hasta los 2 ó 3 años de edad; los niveles de Igs totales se sitúan entre 200 y 400 mg/dL y el número de linfocitos B es normal. Es una ID poco frecuente, con escasas manifestaciones clínicas, que en muchas ocasiones no se diagnostica; afecta por igual a ambos sexos, lo que la diferencia de la ALX. Su incidencia parece ser mayor en familiares de individuos afectados por deficiencia de IgA o inmunodeficiencia variable común.
El paciente debe controlarse hasta que normalice la producción de Igs, y las pautas de vacunación han de retrasarse hasta que exista una respuesta adecuada de Acs, la cual es recomendable comprobar midiendo la tasa de Acs específicos posvacunación. La necesidad de tratamiento con gammaglobulina es excepcional.4
La absorción defectuosa de nutrientes a través de la mucosa intestinal da lugar a un síndrome caracterizado por desnutrición proteico-energética, deposiciones anormales, distensión abdominal y carencias de vitaminas y minerales; en algunos casos, la diarrea puede ser episódica. Cuando la MIS se manifiesta con carácter oligosintomático en forma de un retardo del crecimiento de causa no determinada, y con poca sintomatología digestiva, hace más difícil el diagnóstico. La pérdida entérica de proteínas incluye también la de Igs y de albúmina; sin embargo, a diferencia de la ALX, esta puede afectar a ambos sexos, el número de linfocitos B circulantes es normal y la biopsia de la lámina propia intestinal muestra un número normal de células plasmáticas.12
La AJ es la inflamación sinovial crónica de causa no conocida; se caracteriza por lo general por un cuadro poliarticular que se inicia antes de los 16 años de edad, y que puede comenzar de una manera progresiva con pocos síntomas generales, o hacerlo mediante un cuadro agudo que precede a las manifestaciones artríticas. Otras formas de presentación clínicas son la pauciarticular y la sistémica. Similar a la ALX, en estos pacientes hay detención prematura del crecimiento, pero en este caso, debido a un cierre precoz del cartílago de conjunción. A diferencia, en la AJ las hembras son las más afectadas y son más frecuentes las manifestaciones dérmicas. El nódulo reumatoideo no se halla prácticamente nunca, como es característico encontrar en la artritis del adulto. Los estudios de laboratorio muestran generalmente hipoproteinemia y anemia marcada, el factor reumatoide y los Acs antinucleares son generalmente positivos, la electroforesis de proteína y la cuantificación de Igs muestran hipergammaglobulinemia y aumento de todos los isotipos de Igs respectivamente, contrario a lo observado en la ALX.13
La FQ es un trastorno hereditario multisistémico que afecta a niños y adultos, que se caracteriza principalmente por una obstrucción crónica e infección de las vías respiratorias y por mala digestión y sus consecuencias. A diferencia de la ALX, es frecuente encontrar en estos pacientes pólipos nasales, hipertensión portal, hemorragias por várices esofágicas, pancreatitis recurrente y exantema análogo a la acrodermatitis enteropática. Generalmente se encuentran antecedentes familiares de la enfermedad. Valores de cloruros en sudor iguales o mayores a 60 mEq/L corroboran el diagnóstico clínico. En algunos casos, estas concentraciones son normales; sin embargo, la comprobación de mutaciones específicas y de un fenotipo compatible bastan para confirmar el diagnóstico.14
Bases moleculares
El hecho de que los pacientes con ALX muestran una ausencia o disminución drástica de sus linfocitos B maduros circulantes, y en médula ósea (MO) un número normal de células pre-B, hacía pensar la existencia de un bloqueo en la diferenciación de estas a células maduras inmunocompetentes.15
Investigaciones recientes han mostrado que mutaciones en el gen que codifica para una tirosina quinasa citoplasmática, la que se ha denominado tirosina quinasa de Bruton (Btk), son responsables de la aparición de la ALX en el humano y de la ID ligada al X en el ratón. El gen responsable ha sido identificado en el locus Xq22.16,17
Estudios in vivo e in vitro indican que la proteína Btk es esencial para la supervivencia de la célula B, la progresión del ciclo celular y la proliferación en respuesta a estímulos antigénicos a través de receptores específicos.17,18
Esta proteína muestra una secuencia de aminoácidos (aa) altamente homóloga a los miembros de la familia Src de tirosinas quinasas citoplasmáticas Fyn, Lck y Lyn que fosforilan residuos de tirosina, por lo que están involucradas en la transducción de señales en las células hematopoyéticas. La proteína Btk presenta 4 dominios: SH1, SH2, SH3 y PH. Los dominios SH2 y SH3 están involucrados en el reconocimiento intermolecular y en la regulación de la actividad quinasa (fig.).16,19
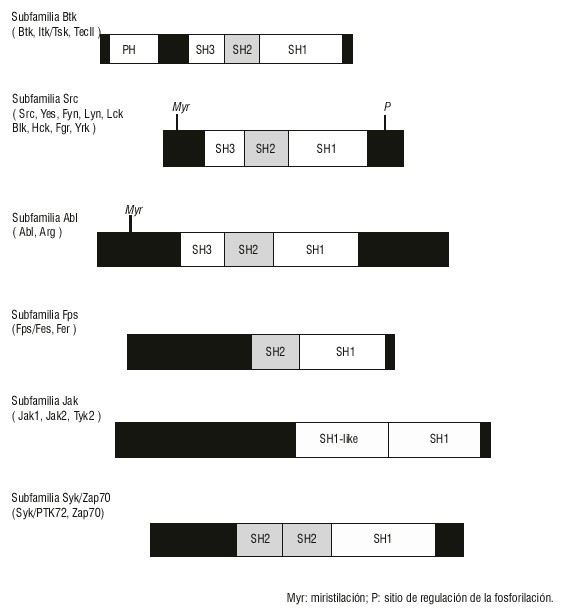
Fig. Comparación de la subfamilia Btk y otras subfamilias de tirosinas quinasas citoplasmáticas.
Las mutaciones de la Btk han sido divididas en 3 categorías: 1) funcionales, cuando afectan los residuos de aa que participan directamente en la unión a fosfotirosinas; 2) estructurales, cuando producen cambios conformacionales que interfieren con el reconocimiento de los residuos de tirosinas fosforilados; y 3) funcioestructurales, cuando incluyen las categorías anteriores. Las principales mutaciones han sido identificadas en los dominios SH2 y SH3.20
La alta heterogeneidad molecular del gen que codifica para la proteína Btk y el grado de expresión de esta, son responsables de la gran variabilidad fenotípica de la enfermedad.21
Recientes estudios han demostrado el papel importante de la Btk en el incremento de las concentraciones intracelulares de calcio en respuesta a estímulos a través del receptor específico de la célula B; la Btk parece estar involucrada en la activación de las proteínas quinasas activadas por estrés JNK/SAPK 2/2 y por lo tanto, en la regulación de c-jun y de otros factores de transcripción importantes en la activación de los genes para citocinas. Esta regulación de la vía de activación de JNK/SAPK puede estar relacionada con la función proapoptótica de la Btk en la muerte celular programada en estas células hematopoyéticas.18,19,22
La proteína Btk se expresa en células pro-B, pre-B, B madura, mastocitos y eritrocitos, no así en células plasmáticas ni en linfocitos T. El hecho de que células mieloides y mastocitos expresen proteínas de la superfamilia de Btk adicionales como Tecll e Itk, pudiera explicar por qué estas células no se encuentran ausentes en pacientes con ALX.23
Estos estudios son la primera demostración de que la mutación o la expresión alterada de una tirosina quinasa citoplasmática puede resultar en la aparición de una enfermedad humana hereditaria; de igual forma, han permitido profundizar en el papel de las tirosinas quinasas en la ontogenia linfoide B y en la patogénesis de las IDP.5-23
Tratamiento
Actualmente el tratamiento de elección de los pacientes con ALX consiste en la administración de gammaglobulina endovenosa a razón de 200-400 mg/kg/dosis cada 3 ó 4 semanas por varios ciclos. Dosis superiores a 500 mg/kg son efectivas para la prevención de infecciones bacterianas severas e insuficiencia pulmonar.24
Otros pilares importantes en el tratamiento de estos pacientes son la educación a familiares acerca de la enfermedad, extremar las medidas de higiene personal y ambiental, la protección de barreras naturales evitando procederes invasivos, la nutrición adecuada, la lactancia materna, así como el uso de antibióticos sistémicos de amplio espectro para el tratamiento de las infecciones. Debe evitarse la administración de vacunas vivas atenuadas.4,24
El trasplante de MO y de stem cell pudieran ser procederes esperanzadores; sin embargo, el uso de la terapia génica, que permite el remplazo del gen afectado, ofrecerá la cura definitiva a estos.25-28
Detección de portadoras y diagnóstico prenatal
Las mujeres heterocigóticas portadoras de ALX pueden ser detectadas por la inactivación al azar del cromosoma X. Las células B de estas mujeres muestran una disminución del crecimiento selectivo en aquellas células que contienen el alelo anormal.29
El diagnóstico prenatal de ALX puede realizarse por la demostración de la ausencia de células B en la sangre fetal.30
La incidencia de las IDP es baja,1,4 sin embargo, su pronóstico muy sombrío,4 por lo que un amplio conocimiento de estas permite diagnosticarlas y tratarlas precozmente, lo que mejora la calidad de vida de los pacientes y disminuye su mortalidad. La naturaleza genética de esta enfermedad posibilita la detección de portadoras asintomáticas, realizar un diagnóstico prenatal con el consiguiente consejo genético para prevenir estos nacimientos y una vía para un mejor entendimiento de la inmunorregulación humana.
Summary
Bruton or X-linked agammaglobulinemia is the prototype of primary B-cell deficiency. Affected male children present with recurrent infections and autoimmune manifestations at the age of 6 months on. The use of modern molecular biology techniques made it possible to detect the gene responsible for the disease in locus Xq22. Genetic character of the disease also allows the detection of carrier mothers and the prenatal diagnosis. Currently, the research on the molecular aspects of the disease continues to be deepened so as to handle the genetic material of patients for therapeutical use, which will result in a final cure for the disease.
Subject headings: AGAMMAGLOBULINEMIA/diagnosis; X CHROMOSOME/immunology; B-LYMPHOCYTES; PROTEIN TYROSINE KINASE/deficiency.
Referencias bibliográficas
1. Ten RM. Primary immunodeficiencies. Mayo Clin Proc 1998;73(9):865-72.
2. Lederman HM. Cancer in children with primary or secondary immunodeficiencies. J Pediatr 1995;127(2):335.
3. García JA, Pacheco A, Regueiro JR. Physiopathogenesis and molecular bases of the primary immunodeficiencies. Sangre 1999;44(2):107-22.
4. Buckeley R. Primary immunodeficiency diseases. En: Paul WE, ed. Fundamental immunology. 3 ed. New York: Raven, 1993:1353-74.
5. Conley ME. X-linked immunodeficiencies. Curr Opin Genet Develop 1994;4:401-6.
6. Rosen FS, Cooper MD, Wedgwood RJ. The primary immunodeficiencies. N Engl J Med 1995;333(7):431-40.
7. Kainulainen L, Varpula M, Liippo K, Svedstrom E, Nikoskelainen J, Ruuskanen O. Pulmonay abnormalities in patients with primary hypogammaglobulinemia. J Allergy Clin Immunol 1999;104(5):1031-6.
8. Fu JL, Shyur SD, Lin HY, Lai YC. X-linked agammaglobulinemia presenting as juvenile chronic arthritis: report of one case. Taiwan Erh Ko I Hsueh Hui Tsa Chih 1999;40(4):280-3.
9. Wang LH, Tsai MJ, Huang MT, Lin SC, Chiang BL. Autoimmune manifestations in pacientes with primary immunodeficiency. Taiwan Erh Ko I Hsueh Hui Tsa Chih 1999;40(4):243-9.
10. Bejaoui M, Barbouche MR, Mellouli F, Tirellil N, Dellagik. Agammaglobulinemia with the absence of circulating B-lymphocytes. 9 cases. Presse Med 1998;27(12):562-7.
11. Wahn V. Evaluation of the child with suspected primary immunodeficiency. Pediatr Allergy Immunol 1995;6(2):71-9.
12. Waldman TA. Protein losing gastroenteropathies. En: Berk JE, ed. Gastroenterology. 4 ed. Philadelphia: Saunder, 1985:1814-5.
13. Bernhard HS. Pediatric rheumatic diseases. En: Schumacher HR, Klippel Th, Koopman WS, eds. Primer on the rheumatic diseases. 10 ed. Atlanta: Arthitis Foundation, 1993:171-6.
14. Boat TF. Fibrosis quística. En: Nelson WE, Berhrman RE, Kliegman RM, Arvin AM, eds. Tratado de Pediatría. La Habana: Editorial Ciencias Médicas, 1977:1554-68.
15. Notarangelo D. Genetics and treatment of primary immunodeficiencies. Introduction. Semin Immunopathol 1998;19(4):363-7.
16. Tsukada S, Rawlings DJ, Witte ON. Role of Bruton´s tyrosine kinase in immunodeficiency. Curr Opin Immunol 1994;6:623-30.
17. Nomura K, Kanegane H, Kartasuyama H, Tsukada S, Agematsu K, Murakami G, et al. Genetic defect in human X-linked agammaglobulinemia impedes a maturational evolution of pro-B cells into a later stage of pre-B cells in the cell differentiation pathway. Blood 2000;96:610-7.
18. Rawling DJ. Bruton´s tyrosine kinase controls a sustained calcium signal essential for B lineage development and function. Clin Immunol 1999;9(13):243-53.
19. Rawling DJ, Witte ON. The Btk subfamily of cytoplasmic tyrosine kinases: structure, regulation and function. Semin Immunol 1995:7(4):237-46.
20. Vihinen M, Kwan SP, Lester T, Ochs HD, Resnick I, Valiaho J, et al. Mutations of the human Btk gene coding for Bruton tyrosine kinase in X-linked agammaglobulinemia. Hum Mutat 1999;13(4):280-5.
21. Gaspar HB, Ferrando M, Caragol I, Hernández M, Bertran JM, De Gracia X, et al. Kinase mutant Btk results in atypical X-linked agammaglobulinemia phenotype. Clin Exp Immunol 2000;120(2):346-50.
22. Petro JB, Rahman SM, Ballard DW, Khan WN. Bruton´s tyrosine kinases is required for activation of kappaB kinase and nuclear factor kappaB in response to B cell receptor engagement. J Exp Med 2000;191(10):17435-54.
23. Kwakami Y, Kitaura J, Hata D, Yao L, Kawakami T. Functions of Bruton´s tyrosine kinase in mast and B cells. J Leukoc Biol 1999;65(3):286-90.
24. Sheaver W, Buckley R, Engles R, Fin A, Fleisher T, Freeman T, et al. Practice parameters for the diagnosis and management. Immunol Allergy 1996;73(3):282-94.
25. Hana I. Immunodeficiencies and possibilites of their modulation. Folia Biol 1996;42(1-2):61-5.
26. Friedrich W. Bone marrow transplantation in immunodeficiency diseases. Ann Med 1996;28(2):115-9.
27. Weinberg KL, Kohn DB. Gene therapy for congenital lymphoid immunodeficiency diseases. Semin Hematol 1998;35(4):354-66.
28. Serrano F, Lain de Lera T, González MA, García MJ, Abad JL, Bernad A, et al. Gene therapy of primary immunodeficiencies. Present and future. Sangre 1999;44(2):143-53.
29. Moschese V, Orlandi P, Plebani A, Arvanitidis K, Florini M, Speletas M, et al. X-chromosome inactivation and mutation pattern in the Bruton´s tyrosine kinase gene in patients with X-linked agammaglobulinemia. Mol Med 2000;6(2):104-13.
30. Curtis SK, Hebert MD, Saha BK. Twin carriers of x-linked agammaglobulinemia due to germline mutation in the Btk gene. Am J Med Genet 2000;90(3):229-32.
Recibido: 20 de octubre del 2000. Aprobado: 4 de enero del 2001.
Dra. Vianed Marsán Suárez. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800, Ciudad de La Habana, Cuba. Teléf: (537)578268. Fax (537) 338979 e-mail:v.marsan@hemato.sld.cu

