










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter v.20 n.2 Ciudad de la Habana Mayo-ago. 2004
Instituto de Hematología e Inmunología
Morbiletalidad en pacientes adultos con drepanocitosis
Dr. Sergio Machín García, Dra. Tamara Guerra Alfonso, Dra. Eva Svarch, Dr. Edgardo Espinosa Martínez, Dr. José R. Mesa Cuervo, Dra. Elvira Dorticós Balea, Dra. Alelí Plasencia Ternblom, Dr. Alejandro González Otero, Dr. Aramís Núñez Quintana y Dra. Maura Wade Mateo
Resumen
Se estudiaron 397 pacientes adultos con drepanocitosis, seguidos entre enero de 1973 y diciembre de 1997; 200 del sexo femenino y 197 del masculino. De estos, 305 con anemia drepanocítica, 63 con hemoglobinopatía SC y 29 con Sb talasemia. La mediana de seguimiento fue de 14,3 años. La media anual de ingresos y transfusiones fue menor en la hemoglobinopatía SC (p<0,001). Las crisis vasooclusivas dolorosas fueron más frecuentes en la Sb talasemia y las infecciones en la anemia drepanocítica (p<0,001). Se desarrollaron un total de 134 embarazos en 87 pacientes. Hubo 4 muertes maternas y 10 muertes perinatales. Se encontraron diferencias en los valores de hemoglobina y reticulocitos entre las hemoglobinopatías (p<0,001), pero no entre los sexos ni grupos de edades (de 18 a 29 años, de 30 a 40 y mayores de 40). La hemoglobina fetal no mostró diferencias significativas entre los sexos y fue mayor en la anemia drepanocítica. La sobrevida global fue de 53 años, en la anemia drepanocítica 53 años, en la hemoglobinopatía SC de 59 y en la Sb talasemia de 48 (p<0,05). Las causas de muerte más frecuentes fueron las crisis hepática, accidentes vasculares encefálicos y las infecciones.
Palabras clave: drepanocitosis, morbilidad.
La drepanocitosis comprende un grupo de anemias hemolíticas crónicas hereditaias en las que está presente el gen de la hemoglobina (Hb) S. Este gen está ampliamente difundido en África y fue trasladado a América mediante el comercio de esclavos. En Cuba se considera un problema de salud pública. La frecuencia del estado de portador AS es de 3,08 % en la población general. 1
La anemia drepanocítica (AD) o hemoglobinopatía SS es la de mayor incidencia; le siguen en orden de frecuencia la hemoglobinopatía SC (HSC) y la Sb talasemia (Sbtal) que puede ser SbOtal o Sb+tal.
Las manifestaciones clínicas más comunes de estas entidades son: anemia, ictericia, crisis vasooclusivas (CVO) dolorosas recurrentes e infecciones bacterianas. En el niño se puede producir el síndrome mano-pie y una causa importante de morbilidad y también de mortalidad es la crisis de secuestro esplénico. El síndrome torácico agudo (STA) es un motivo muy frecuente de ingreso y la complicación más grave es el accidente vascular encefálico (AVE) de tipo oclusivo o hemorrágico. Otras manifestaciones son: priapismo, úlceras maleolares, necrosis aséptica de la cabeza de los huesos largos, retinopatía proliferativa y litiasis vesicular.1,2
También existe una oclusión microvascular subclínica que conduce en el paciente adulto a un daño orgánico crónico sobre todo al nivel pulmonar, cardíaco y renal. 3
Aunque se ha avanzado mucho en el conocimiento de la fisiopatología de la drepanocitosis en las últimas décadas, todavía no se conoce completamente su historia natural. Por otra parte, desde diferentes partes del mundo se comunican diferencias en la expresión clínica de la enfermedad. 2
El propósito de este trabajo es describir la evolución de un grupo numeroso de pacientes con drepanocitosis seguidos durante un período de tiempo prolongado en Instituto de Hematología e Inmunología.
Métodos
Se realizó un estudio retrospectivo de 397 enfermos adultos que fueron vistos regularmente en consulta externa cada 3 - 6 meses durante el período enero 1973 a diciembre 1997. En cada consulta se realizó interrogatorio para registrar los episodios ocurridos entre las consultas. También se practicó el examen físico, Hb y reticulocitos por las técnicas habituales; sus valores se compararon entre grupos de edades: de 18 a 29 años, de 30 a 40 y más de 40, por sexos y por tipo de hemoglobinopatía.
La Hb fetal (HbF)4 se determinó en 287 pacientes en algún momento de su evolución.
Se calculó el promedio anual de ingresos, transfusiones, infecciones y CVO dolorosas. Estas últimas se definieron como la presencia de dolor osteomioarticular o abdominal de más de 6 horas de duración, sin causa demostrable, y las infecciones por la presencia de fiebre de más de 7 días de evolución, aún sin signos de localización o cualquier proceso infeccioso demostrable.
Se determinó la frecuencia de otras manifestaciones clínicas durante todo el período.
El STA se definió como la aparición de manifestaciones respiratorias con presencia de nuevas lesiones de aspecto inflamatorio en la radiografía de tórax; la crisis hepática por la intensificación de la ictericia con hepatomegalia dolorosa a la palpación y elevación de las enzimas hepáticas; la crisis de secuestro hepático, cuando se observó una caída de la Hb de 2 g/dL o más, con aumento de los reticulocitos y aumento doloroso del tamaño del hígado; la crisis hepática mixta cuando se presentaron intensificación de la ictericia, además de los criterios de secuestro hepático; y la pancreatitis por dolor abdominal y vómitos con valores de amilasa sérica elevados.
Las causas de muerte en la mayoría de los enfermos se tomaron de los certificados de defunción y por los informes anatomopatológicos en 38 pacientes en que se realizó necropsia.
Todos los datos clínicos y de laboratorio se procesaron por separado en cada una de las hemoglobinopatías, aunque no fue posible en la Sbotal y la Sb+tal por el pequeño número de casos.
La sobrevida global de acuerdo con el tipo de hemoglobinopatía se determinó por el método de Kaplan-Meier. Para el análisis de variables separadas se utilizó la prueba de Chi cuadrado y para los datos continuos se compararon los valores promedios con el análisis de varianza y de regresión lineal. El nivel de significación fue del 5 % en todos los casos.
Resultados
La distribución de los pacientes por edad, sexo y hemoglobinopatía se muestra en la tabla 1.
TABLA 1. Distribución por sexo y hemoglobinopatía
| Hemoglobinopatía | ||||
| Sexo | AD | HSC | Sb/tal | Total |
| Femenino | 151 | 38 | 11 | 200 |
| Masculino | 154 | 25 | 18 | 197 |
| Total | 305 | 63 | 29 | 397 |
Sb0/tal: 22 pacientes.
Sb+/tal: 7 pacientes.
La mediana de seguimiento fue de 14,3 años (3-25).
El promedio anual de CVO dolorosas, infecciones, hospitalizaciones y transfusiones se observa en la tabla 2. Las hospitalizaciones y transfusiones fueron menos numerosos en la HSC (p< 0,001). Las CVO dolorosas fueron más frecuentes en la Sb tal y las infecciones en los pacientes con AD (p< 0,001).
TABLA 2. Promedio anual de ingresos, CVO dolorosas, infecciones y transfusiones
según hemoglobinopatía
| Hemoglobinopatía | ||||||
| Manifestación | AD | HSC | Sb/tal | |||
| X | De | X | De | X | DE | |
| Ingresos | 0,4 | 0,7 | 0,14* | 0,4 | 0,32 | 0,7 |
| CVO dolorosas | 0,8 | 1,4 | 0,85 | 1,4 | 1,02* | 1,4 |
| Infecciones | 0,24* | 0,5 | 0,14 | 0,4 | 0,12 | 0,4 |
| Transfusiones | 0,3 | 1,1 | 0,07* | 0,6 | 0,22 | 0,9 |
* p < 0,001.
En la tabla 3 se expone la incidencia de otras manifestaciones clínicas. El STA, la crisis hepática y la litiasis vesicular se observaron menos en la HSC (p<0,05). La pancreatitis se asoció en todos los casos con la crisis hepática. El AVE fue hemorrágico en el 22,5 % de los enfermos.
TABLA 3. Otras manifestaciones clínicas por hemoglobinopatías
| Hemoglobinopatía | ||||||||
| Evento | AD | HSC | Sb/tal | Total | ||||
| No. | % | No. | % | No. | % | No. | % | |
| Síndrome torácico agudo* | 106 | 34,8 | 13 | 20,6 | 13 | 44,8 | 132 | 33,2 |
| Crisis hepática* | 87 | 29 | 2 | 3,2 | 6 | 20.7 | 95 | 23,9 |
| Litiasis vesicular** | 100 | 32,8 | 10 | 15,9 | 2 | 6,9 | 112 | 28,2 |
| Úlcera maleolar | 29 | 9,5 | 3 | 4,8 | 0 | 0 | 32 | 8,1 |
| Accidente vascular encefálico (AVE) | 23 | 7,5 | 3 | 4,8 | 5 | 17,2 | 31 | 7,8 |
| Necrosis aséptica del fémur | 24 | 7,9 | 3 | 4,8 | 0 | 0 | 27 | 6,8 |
| Insuficiencia renal crónica | 18 | 5,9 | 1 | 1,6 | 0 | 0 | 19 | 4,8 |
| Crisis de secuestro hepático | 16 | 5,2 | 0 | 0 | 0 | 0 | 16 | 4 |
| Crisis hepática mixta | 10 | 3,3 | 1 | 1.6 | 0 | 0 | 11 | 2,8 |
| Pancreatitis | 7 | 2,3 | 0 | 0 | 1 | 3,4 | 8 | 2 |
* p <0,05 **, p <0,001.
El promedio anual de los valores de Hb, reticulocitos y HbF se describe en la tabla 4. Se encontraron diferencias significativas en los valores de hemoglobina y reticulocitos entre las hemoglobinopatías (p< 0,001), no así en las cifras de HbF. No se observaron diferencias significativas en la hemoglobina y los reticulocitos entre edades y sexos, ni en la HbF entre los sexos.
TABLA 4. Promedio anual de los parámetros hematológicos por hemoglobinopatías
| Hemoglobinopatía | ||||||
| Parámetro | AD | HSC | Sb/tal | |||
| X | DE | X | DE | X | DE | |
| Hemoglobina (g/dL)* | 7,6 | 2,2 | 11 | 2,9 | 8,7 | 2,3 |
| Reticulocitos(%)* | 12 | 5 | 6,2 | 2,9 | 8,7 | 4,6 |
| Hb F (%) | 9 | 9,3 | 3,8 | 3,4 | 6,6 | 4,3 |
* p <0,001.
Ocurrieron 134 embarazos en 87 mujeres (43,5 %). Hubo 49 interrupciones: 17 abortos espontáneos, 29 provocados (10 por complicaciones de la madre o el feto) y 3 embarazos ectópicos rotos. Llegaron a término 85 embarazos. Las muertes perinatales fueron 7 fetales y 3 recién nacidos, que representan 11,7 % del total de embarazos que llegaron a término. Hubo 4 muertes maternas, todas en pacientes con AD.
Se encontró una sobrevida global de 53 años en al AD; 59 en la HSC y 48 en la Sbtal (fig. 1). Hubo diferencias significativas entre las hemoglobinopatías (p< 0,05), pero no entre los sexos (p=0,11) (fig. 2).
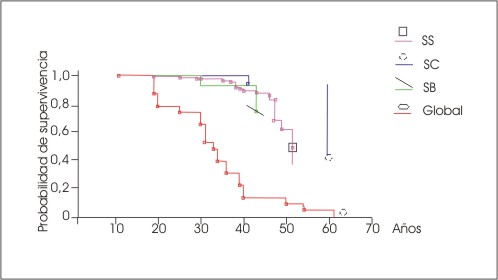
FIG. 1 Sobrevida según hemoglobinopatía.
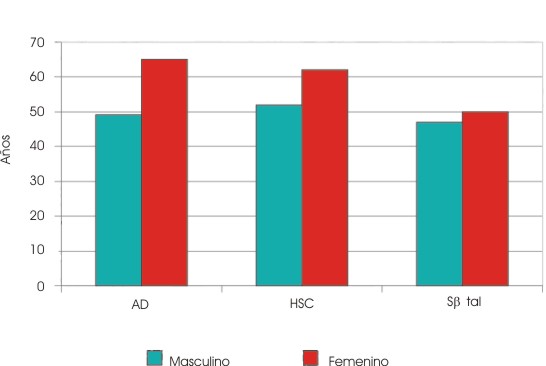
FIG. 2 Sobrevida según hemoglobinopatías y sexo.
AD: anemia drepanocítica; HSC: hemoglobinopatía SC; Sbtal: Sb talasemia
Se determinaron como causas más frecuentes de muerte las complicaciones hepáticas, el AVE y las infecciones (tabla 5).
TABLA 5. Causas de muerte por hemoglobinopatías
| Hemoglobinopatía | ||||||||
| Causas | AD | HSC | Sb/tal | Total | ||||
| No. | % | No. | % | No. | % | No. | % | |
| Crisis hepática | 12 | 23,5 | 0 | 0 | 0 | 0 | 12 | 20,7 |
| Crisis del SNC | 9 | 17,6 | 0 | 0 | 0 | 0 | 9 | 15,5 |
| Infección | 4 | 7 8 | 1 | 25 | 1 | 33 3 | 6 | 10,4 |
| Desconocida | 7 | 13,7 | 0 | 0 | 1 | 33,3 | 8 | 13,8 |
| Síndrome torácico agudo | 5 | 9,8 | 0 | 0 | 0 | 0 | 5 | 8,6 |
| Secuestro masivo | 5 | 9,8 | 0 | 0 | 0 | 0 | 5 | 8,6 |
| Insuficiencia cardíaca | 4 | 7,8 | 1 | 25 | 0 | 0 | 5 | 8,6 |
| Otras causas | 3 | 5,9 | 0 | 0 | 1 | 33,3 | 4 | 6,9 |
| Infarto de médula ósea | 1 | 2 | 2 | 50 | 0 | 0 | 3 | 5,2 |
| Reacción hemolítica grave | 1 | 2 | 0 | 0 | 0 | 0 | 1 | 1,7 |
| Total | 51 | 87,9 | 4 | 6,9 | 3 | 5,2 | 58 | 100 |
Discusión
Las diferencias clínicas y hematológicas entre la AD y la HSC son bien conocidas. 1,2
No es posible en este trabajo interpretar la mayor incidencia de CVO dolorosas en la Sbtal, ya que el número de enfermos estudiado fue pequeño y no se separó la SbOtal y la Sb+tal. Es conocido que la Sb0tal tiene un cuadro clínico semejante a la AD y la Sb+tal a la HSC. 1,5 También es difícil la comparación de la incidencia de STA en el presente estudio con los datos de la literatura, ya que la definición de este síndrome es posterior al inicio del seguimiento de los enfermos; por otro lado, aún hoy existen diferentes criterios para su diagnóstico. 6-8
En la drepanocitosis se describen lesiones hepáticas producidas por falciformación crónica en los sinusoides, hepatitis viral, sobrecarga de hierro o una combinación de estos factores. 1 La crisis hepática que posiblemente se produce por una vasooclusión intrahepática aguda, fue descrita por primera vez por Diggs en 1965. 9 Posteriormente se demostró que su curso clínico es muy variable desde la resolución espontánea en poco días hasta la insuficiencia hepática y la muerte. 2,10 Es posible que inmunocomplejos circulantes participen en su patogénesis, ya que un paciente mejoró rápidamente después de una plasmaféresis. 11,12 La alta frecuencia de esta complicación en nuestro medio no tiene una explicación clara. En un estudio realizado en nuestra institución, donde se practicó biopsia a 50 pacientes, no se pudo demostrar hepatitis viral. 13
La litiasis vesicular tuvo una incidencia similar a la encontrada en Estados Unidos de Norteamérica 14 y menor que la que se comunica en Jamaica. 2
La frecuencia de AVE fue similar a lo descrito en otros estudios. 2,15
La insuficiencia renal crónica puede presentarse entre el 4 y 18 % de los adultos con AD; 16 en nuestros pacientes se diagnosticó en el 5,1 %.
Es interesante señalar la menor incidencia de úlceras maleolares en este estudio. 17 Es posible que las medidas profilácticas que se utilizan (máxima higiene, uso de medias y calzado bajo), hayan influido en este hallazgo.
La evolución del embarazo en nuestras pacientes fue muy buena. A partir de 1988 no falleció ninguna enferma. La mortalidad perinatal también fue baja. Las causas que conducen a la muerte de la madre o el niño son la toxemia gravídica, el STA, las crisis hepáticas y las complicaciones del parto. 18
Se encontraron diferencias significativas de los parámetros hematológicos entre la AD y la HSC, tal como se ha señalado. 1,2 Se ha reconocido que existen variaciones en los valores de Hb entre los diferentes grupos de edades y los sexos. En el grupo de 10 a 24 años los valores de Hb son significativamente más altos en el sexo masculino, y después de los 40 años disminuyen en ambos sexos. 19 En nuestro estudio se encontraron cifras mayores de hemoglobina en los hombres, pero las diferencias no fueron significativas. La razón por la que no se demostró una disminución de la hemoglobina en los enfermos de más de 40 años no está clara.
La HbF no tuvo diferencias significativas entre las hemoglobinopatías ni los sexos, a pesar de que fue más alta en las mujeres tal como se describe, 2 pero no se estudiaron todos los pacientes.
La sobrevida en los enfermos en este estudio es mayor en la AD, similar en la HSC a la encontrada en algunos trabajos 3,20 superior a lo descrito en otros. 21 No hubo diferencias significativas entre los sexos, aunque el promedio de vida de las mujeres fue mayor, hallazgo ya referido en la literatura. 22 Nuestro grupo incluye mucho menos enfermos y además describe un grupo seleccionado con fácil acceso al hospital, y atención médica especializada por un equipo entrenado.
Las causas más frecuentes de muerte descritas en la literatura son el STA, la insuficiencia renal crónica, las infecciones y los AVE. 2,3,23,24 La primera causa de muerte en nuestro estudio fueron las complicaciones hepáticas, para las que no tenemos explicación clara.
La mayoría de nuestros enfermos falleció durante un episodio agudo. Sólo el 17 % murió por un daño orgánico crónico, lo que está de acuerdo con lo descrito en otros estudios. 3,23
Este trabajo describe el cuadro clínico, algunos parámetros hematológicos y la sobrevida en un grupo de pacientes que recibieron una atención médica integral, lo que se refleja en una alta sobrevida.
Summary
A study was performed on 397 adult patients with sickle cell anemia, who had been followed up from January 1973 to December 1997. Two hundred were females and 197 males; 305 of these patients presented with sickle cell anemia, 63 with SC hemoglobinopathy and 29 with Sb thalassemia. The mean follow-up period was 14,3 years. The yearly admission and blood transfusion mean was lower in SC hemoglobinopathy(p<0,001). Painful vasoocclusive crisis was more frequent in Sb thalassemia whereas infections often occurred in sickle cell anemia (p<0,001). One hundred four pregnancies were developed from 87 females. There were 4 maternal deaths and 10 perinatal deaths. Differences were found in hemoglobin and reticulocyte values among hemoglobinopathies (p<0,001) but no difference was seen between sexes or among age groups (18-29 y, 30-40 y and over 40 y). Fetal hemoglobin values did not show significant difference between sexes, although they were higher in sickle cell anemia. Global survival estimates were 53 years in sickle cell anemia, 59 years in SC hemoglobinopathy and 48 in Sb thalassemia (p< 0,05). The most frequent cases of death were hepatic complications, encephalic vascular attacks and infections.
Key words: sickle cell anemia, morbidity
Referencias bibliográficas
- Colombo B, Svarch EG, Martínez G. Genética y clínica de las hemoglobinopatías humanas. Ciudad de La Habana: Pueblo y Educación; 1994. p. 146-95.
- Serjeant GR, Serjeant BC. Sickle cell disease. 3 ed. Oxford: Oxford University; 2001.
- Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al . Mortality in sickle cell disease : Life expectancy and risk factor for early death. N Engl J Med 1994;330:1639-44.
- Betke K, Marti H, Schicht I. Estimation of small percentages of faetal haemoglobin. Nature 1959;184:1877-84.
- Farber MD, Koshy M, Kinney TR and the Cooperative Study of Sickle Cell Disease. Cooperative study of sickle cell disease: demographic and socioeconomic characteristics of patients and families with-sickle cell disease. J Chron Dis 1985;38:495-505.
- Platt OS. The acute chest syndrome of sickle cell disease. N Eng J Med 2000;342:1904-7.
- Quinn CT, Buchanan GR. The acute chest syndrome of sickle cell disease. J Pediatr 1999;135:416-22.
- Ballas SK. Acute chest syndrome in sickle cell anemia. Intensive care med 2000;15:123-5.
- Diggs LW. Sickle cell crisis. Am J clin Pathol 1965;44:1-19.
- Sheehy TW. Sickle cell hepatopathy. Southern Med J 1977;70:533-8.
- Svarch E, González A, Villascusa R, et al. Plasma exchange for acute cholestasis in homozygous sickle cell disease. Haematologia 1986;19:49-51.
- Hernández P, Carnot J, Cruz C, Dorticós E, Espinosa E, Villaescusa R, et al. Circulating immune complexes in sickle cell hepatic crisis. Acta Haematol 1981;65:15-20.
- Espinosa E, Barreto G, Cabrera J, Hernández P. El hígado en la anemia drepanocítica. Un estudio clínico patológico en 50 pacientes. Sangre 1985;30:945-50.
- Schubert TT. Hepatobiliory system in sickle cell disease. Gastroenterology 1986;90:2013-21.
- Powars D. Sickle cell anemia and major organ failure. Hemoglobin 1990;14:573-98.
- Eckman R. Leg ulcers in sickle cell disease. Hematol Oncol Clin North Am 1996;10:1333-44.
- Falk RJ, Jeannette JC. Sickle cell nephropathy. Adv Nephrol 1994;23:133-47.
- Koshy M. Sickle cell disease and Pregnancy. Blood Rev 1995;9:157-64.
- Hayes RJ, Beckford M, Grandison Y, Mason K, Serjeant BE, Serjeant GR. The haematology of steady state homozygous sickle cell disease ; frequency distributions variation with age and sex, longitudinal observations. Br J Haematol 1985;59:369-82.
- Wierenga KJ, Hambleton IR, Lewis NA. Survival estimates for patients with sickle-cell disease in Jamaica: a clinc-based population study. Lancet 2001;139:680-3.
- Powars DR, Hiti A, Ramicone E, Johnson C, Chan L. Outcome in hemoglobin SC disease: a four-decade observational stidy of clinical, hematologic, and genetic factors. Am J Hematol 2002;70:206-15.
- Kotila TR, Shokunbi WA. Survival advantage in female patients with sickle cell anaemia. East Afr Med J 2001;78:373-5.
- Perronne V, Roberts-Harewood M, Bachir D, Roudot-Thoraval F, Delerod JM, Thuret I, et al. Patterns of mortality in sickle cell disease in adults in France and England. Hematol J 2002;3:56-60.
- Embury SH. Sickle cell disease. En: Hoffman R, Benz GJ, Shattil SJ, Furie B, Cohen HJ, Silberstein LE, eds. Hematology Basic principles and practice. New York: Churchill Livingstone; 1995. p. 611-48.
Recibido: 6 de junio del 2004. Aprobado: 2 de julio del 2004.
Dr. Sergio Machín García. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800, Ciudad de La Habana, Cuba. Tel (537) 578268, 578695, 544214. Fax (537) 442334. e-mail: ihidir@hemato.sld.cu

