










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión impresa ISSN 0864-0289
Rev Cubana Hematol Inmunol Hemoter vol.27 no.4 Ciudad de la Habana oct.-dic. 2011
PRESENTACIÓN DE CASOS
Síndrome Beguez-Chediak-Higashi. Comunicación de un nuevo caso en Cuba
Beguez Chediak-Higashi Syndrome. Report of a new case in Cuba
Dr. Sergio Machín García, Prof. DraC. Eva Svarch, Dr. Alejandro González Otero, Dra. Andrea Menéndez Veitía, Dra. Ania Hernández Cabezas, Dr. Jesús Serrano Mirabal, Dr. Alberto Arencibia Núñez, Dra. Addys Gutiérrez Díaz
Instituto de Hematología e Inmunología. La Habana, Cuba.
RESUMEN
El síndrome de Beguez-Chediak-Higashi es una enfermedad rara, autosómica recesiva, descrita en Cuba por el Dr. Beguez-César en 1943. Se presenta un paciente masculino de 8 meses de edad con antecedentes de infecciones graves, obesidad, palidez cutáneo-mucosa intensa, cabello de color plateado, hepatoesplenomegalia y anemia con presencia de gránulos lisosomales gigantes en las células del sistema granulopoyético. Se trató con prednisona, vincristina, etopósido y aciclovir oral con respuestas parciales y transitorias.
Palabras clave: Beguez-Chediak-Higashi.
ABSTRACT
Beguez-Chediak-Higashi syndrome is a rare illness; it was described in 1943 by Dr. Beguez-Cesar in Cuba. We present an 8 months boy with frequent infections, obesity, intense paleness, silver hair, hepatomegaly, splenomegaly, anemia and big lisosomal granules in myelopoietic system. He was treated with prednisolona, vincristin, VP-16, and oral acyclovir with partial and transitory results.
Key words: Beguez-Chediak-Higashi.
INTRODUCCIÓN
El síndrome de Beguez-Chediak-Higashi es una enfermedad rara, autosómica recesiva, que fue descrita por primera vez en Cuba en 1943, por el Dr. Beguez-Cesar en una familia de 13 hermanos de los cuales 4 eran enfermos.1 La alteración genética está relacionada con mutaciones en el gen CHS1/LYST en el cromosoma 1q.2-5
Se caracteriza por albinismo óculo-cutáneo parcial, fotofobia, nistagmus, alteraciones de la coagulación, manifestaciones neurológicas variables y alteraciones de la inmunidad, sobre todo de las células citotóxicas naturales, que predisponen a infecciones virales y bacterianas.6-12 El diagnóstico se confirma por la presencia de gránulos citoplasmáticos gigantes en los leucocitos. El 85 % de los pacientes desarrollan una fase acelerada en algún momento de su evolución, con características clínicas y hematológicas muy similares a las de la linfohistiocitosis hemofagocítica.13 La infección por el virus de Epstein-Barr se ha descrito como desencadenante de esta fase de la enfermedad, aunque no ha sido totalmente probado.6,14
El trasplante de células progenitoras hematopoyéticas es el tratamiento de elección, efectivo en los trastornos hematopoyéticos e inmunológicos, pero ineficaz en las manifestaciones neurológicas.15,16 El tratamiento actual de la fase acelerada es el protocolo de la linfohistiocitosis hemofagocítica (LNH-94).17
PRESENTACIÓN DEL CASO
Paciente masculino de 8 meses de edad, hijo de padres sanos no relacionados, con 2 hermanos gemelares univitelinos normales, que presentó infecciones graves y muchos episodios febriles de causa no precisada.
Al examen físico se encontró obesidad, intensa palidez cutáneo-mucosa, cabello de aspecto plateado, hígado y bazo que rebasaban el reborde costal en 1 y 4 cm, respectivamente, y retraso del desarrollo psicomotor.
En el fondo de ojo se observaron los vasos coroides por alteraciones de la capa pigmentaria de la retina.
Los estudios de laboratorio mostraron: hemoglobina 78 g/L, reticulocitos 11 %, leucocitos 5 ´ 109/L, neutrófilos 17 %, linfocitos 81 %, monocitos 2 % y plaquetas 260 ´ 109/L. La velocidad de eritrosedimentación fue de 87 mm/h. En las extensiones de sangre periférica y médula ósea se observaron muchos gránulos lisosomales gigantes en las células del sistema granulopoyético y en los linfocitos (fig. 1). Los restantes complementarios fueron normales: alaninoaminotransferasa, urea, creatinina, hierro sérico, ácido fólico, vitamina B12, muramidasa en plasma, prueba de Coombs y los títulos de anticuerpos contra el antígeno capsular del virus de Epstein Barr.
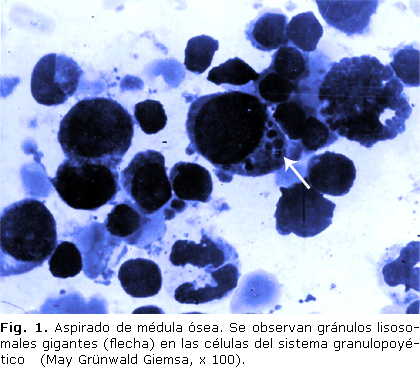
Los estudios imagenológicos mostraron esplenomegalia ligera en la ultrasonografía abdominal y en la tomografía axial computadorizada del sistema nervioso central se observaron: atrofia cortical, cisterna magna amplia, dilatación del cuarto ventrículo y atrofia cerebelosa severa.
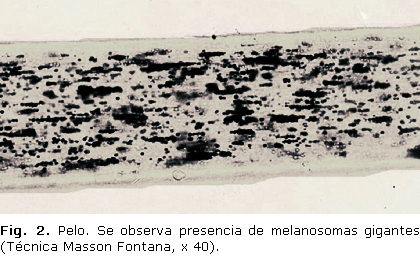
La biopsia de cabello mostró melanosomas gigantes (fig. 2).
Se consideró al paciente en fase acelerada de la enfermedad y se inició tratamiento con prednisona (2 mg/kg/día), vincristina (1,5 mg/m2 semanal 4 dosis) y aciclovir oral (20 mg/kg/día) por 21 días. La fiebre desapareció, disminuyó la visceromegalia y se normalizó la hemoglobina. Dos meses después reaparecieron las manifestaciones clínicas y de laboratorio, por lo que se repitió igual tratamiento con buena respuesta, pero transitoria. La adición de etopósido (150 mg/m2/día, 3 dosis) produjo una respuesta parcial durante un mes. El niño falleció en el curso de una sepsis generalizada.
El estudio anátomo-patológico mostró, además de las alteraciones propias de la enfermedad, una bronconeumonía bilateral extensa, miocarditis intersticial, enteritis aguda, peritonitis focal, esteatosis hepática severa, infartos hemorrágicos esplénicos, hipocelularidad de la médula ósea con depresión de las 3 líneas hematopoyéticas.
DISCUSIÓN
La mayoría de los pacientes con este síndrome en los que no se realiza el trasplante de células hematopoyéticas, mueren antes de los 10 años de edad, fundamentalmente por infecciones.10,11 La muerte puede ser debida también a la fase acelerada que es muy semejante a la linfohistiocitosis hemofagocítica y que para algunos autores es desencadenada por la infección del virus de Epstein Barr.6,13,14 En nuestro paciente hubo un progresivo agravamiento clínico y hematológico, que no se pudo atribuir a una infección por este virus.
Durante su evolución se observó reticulocitosis sin evidencias de anemia hemolítica autoinmune. Los valores más elevados de reticulocitos y la anemia intensa se presentaron asociados con manifestaciones clínicas de la fase acelerada. Hasta donde conocemos, estas alteraciones no se han descrito en la literatura.5,13 Aunque no se pudo demostrar, es probable que existiera algún grado de hiperesplenismo. La hipocelularidad de la médula ósea descrita en la necropsia se interpretó como secundaria a los tratamientos utilizados.
En el presente caso hubo respuesta de corta duración a la quimioterapia utilizada que coincide con lo que han encontrado otros autores.5,13 Hasta el momento, el trasplante de células progenitoras hematopoyéticas es el único tratamiento curativo de esta enfermedad.15,16
El Dr. Béguez-César fue reconocido mundialmente como el descubridor de esta enfermedad, durante la I Jornada Latinoamericana de Estudios Cooperativos en Hematología, celebrada en La Habana en 1973.18 Aunque la enfermedad haya sido más comúnmente divulgada con el nombre de síndrome de Chediak-Higashi, actualmente ya es reconocida en la literatura internacional como síndrome de Beguez-Cesar-Chediak-Higashi.19,20
REFERENCIAS BIBLIOGRÁFICAS
1. Béguez-Cesar AB. Neutropenia crónica maligna familiar con granulaciones atípicas de los leucocitos. Bol Soc Cubana Pediatr. 1943;15:900-22.
2. Barrat FG, Auloge L, Postural E, Logelouse RD, Velmer E, Cant AJ, et al. Genetic and physical mapping of the Chediak-Higashi sindrome on cromosoma 1q 42-43. Am J Hum Genet. 1996;59:625-32.
3. Ward DM, Sheflett SL, Kaplan J. Chediak-Higashi syndrome. Curr Opin Hematol. 2008;15:22-9.
4. Zarzour W, Kleta R, Frangoul H, Suwannarat P, Jeong A, Kim SY, et al. Two novel CHS1(LYST) mutations: clinical correlations in an infant with Chediak-Higashi syndrome. Mol Genet Metab. 2005;85:125-32.
5. Kaplan J, De Domenico I, Ward DM. Chediak-Higashi syndrome. Curr Opin Hematol. 2008;15:22-9.
6. Introne W, Westbroek W, Golas GA, Adams D. Chediak-Higashi syndrome. Disponible en: http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=chediak-higashi
7. Buchanan GR, Handin RL. Platelets function in Chediak-Higashi syndrome. Blood. 1976;47:941-8.
8. Sung JH, Stadian EM. Neurophatological changes in Chediak-Higashi disease. J Neurophatol Exp Neurol. 1968;27:156-7.
9. Tardieu M, Lacroix C, Neven B, Bordigoni P, de Saint Basile G, Blanche S, et al. Progressive neurologic dysfunction 20 years after allogeneic bone marrow transplantation for Chediak-Higashi syndrome. Blood. 2005;106:40-2.
10. Padgett GA, Geiqnam CW, Henson JB, Gorham JR. Comparative studies of susceptibility of infection in the Chediak-Higashi syndrome. J Pathol Bacteriol. 1968;95:509-22.
11. Welkenson PC. Defect of leukocyte locomotion and chemotaxis: prospects, assays and lessons from Chediak-Higashi neutrophils. Eur J Clin Invest. 1993;23:690-2.
12. Bailleul-Forestier I, Manod-Broca J, Benkerrou M, Mora F, Picard B. Generalized periodontitis associated with Chediak-Higashi syndrome. J Periodontol. 2008;79:1263-70.
13. Bejaoni M, Veber F, Gerault D, Gaud C, Blanche S, Gricelli C, et al. The accelerated phase of Chediak-Higashi syndrome. Arch Fr Pediatr. 1989;46:733-6.
14. Merino F, Henle W, Ramírez-Duque P. Chronic active Epstein-Barr virus infection in patients with the Chediak-Higashi syndrome. J Clin Immunol. 1986;6:299-305.
15. Haddad E, Le Deist F, Blanche S, Benkerrou M, Rohrlich P, Vilmer E, et al. Treatment of Chediak-Higashi syndrome by allogenic bone marrow transplantation: report of 10 cases. Blood. 1995;85:3328-33.
16. Eapen M, DeLaat CA, Baker KS, Cairo MS, Cowan MJ, Kurtzberg J, et al. Hematopoietic cell transplantation for Chediak-Higashi syndrome. Bone Marrow Transplantation. 2007;39:4115.
17. Henter JI, Samuelsson-Horne A, Aricò M, Egeler RM, Elinder G, Filipovich AH, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood 2002;100:236773.
18. Peña Sánchez MA, Miyares Peña MV, Batista Duharte A, Paúltre Rivas S, Béguez López V. Antonio Béguez César y su descubrimiento de la neutropenia crónica maligna familiar con granulaciones atípicas de los leucocitos [artículo en línea]. MEDISAN 2007;11(3). [citado 23 mayo 2011]. Disponible en: http://bvs.sld.cu/revistas/san/vol11_3_07/san16307.htm
19. Zabala P, Fontán G, Lorente F, Kreisler M, Sanjuán I, Barbolla L, et al. Enfermedad Beguez-Cesar-Chediak-Higashi: estudio de un caso en fase de seudo-linfoma. Sangre. 1982;27(1):88-95.
20. Babylon.com [Internet]. Diccionario online. Medciclopedia. Disponible en: http://diccionario.babylon.com/s%C3%ADndrome%20de%20beguez-cesar-steinbrinck-chediak-higashi/
Recibido: 7 de junio de 2011.
Aprobado: 25 de junio de 2011.
Dr. Sergio Machín García. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800, La Habana, Cuba. Tel (537) 643 8695, 8268, Fax (537) 644 2334. Correo electrónico: ihidir@hemato.sld.cu
Website: http://www.sld.cu/sitios/ihi

