










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión impresa ISSN 0864-0289
Rev Cubana Hematol Inmunol Hemoter vol.29 no.3 Ciudad de la Habana jul-sep. 2013
REPORTE DE CASO
Atrofia hemifacial progresiva o Síndrome Parry Romberg asociado a inmunodeficiencia
Progressive Hemifacial Atrophy or Parry Romberg Syndrome associated with immunodeficiency
Dra. Miriam de la C. Sánchez Segura, Dra. Vianed Marsán Suárez, Dra. Consuelo Macías Abraham, Dra. Alina García García, Dra. Julio Valcárcel Llerandi, Lic. Lázaro O. del Valle Pérez, Lic. Beatriz Socarrás Ferrer, Lic. Ada A. Arce Hernández
Instituto de Hematología e Inmunología. La Habana, Cuba.
RESUMEN
La Atrofia Hemifacial Progresiva (AHP) o Síndrome Parry Romberg, es una enfermedad degenerativa rara, caracterizada por una lenta y progresiva atrofia facial unilateral que afecta al tejido celular subcutáneo, cartílago, tejido graso y estructuras óseas subyacentes, que frecuentemente se solapa con una condición conocida como esclerodermia lineal «en corte de sable». Hasta donde se conoce no se ha reportado en la literatura la asociación de este síndrome a algún tipo de inmunodeficiencia. Se presenta el caso de un niño de 5 años con AHP, con historia de procesos infecciosos recurrentes, algunos graves, desde que tenía 7 meses de nacido. En el estudio inmunológico se observó la presencia de anticuerpos antinucleares con patrón homogéneo y de anticuerpos anti-DNA de doble cadena. La cuantificación de las subpoblaciones linfocitarias mostró una disminución de los valores de células T/CD3+ y T/CD4+, con valor normal de células B/CD19+. Se diagnosticó una inmunodeficiencia de células T. El hallazgo de una inmunodeficiencia celular en un paciente con AHP es expresión de la gran variabilidad clínica de esta enfermedad y de la importancia que tiene su diagnóstico temprano.
Palabras clave: inmunodeficiencias, Síndrome Parry Romberg, Atrofia hemifacial Progresiva.
ABSTRACT
The progressive hemifacial atrophy (AHP) or Parry Romberg syndrome, is a rare degenerative disease, characterized by slowly progressive unilateral facial atrophy involving the subcutaneous tissue, cartilage, fat tissue and underlying bone structures, which often overlaps with a condition known as linear scleroderma «en coup of sabre». To our knowledge has not been reported the association between immunodeficiency and this syndrome. We report the case of a child of 5 years with AHP, with a history of recurrent infectious processes, some serious, since he was 7 months old. The immunological study showed T cell immunodeficiency, lymphocyte subpopulations showed T/CD4 T/CD3 + cells values decreased and normal value B/CD19 + cells. The presence of antinuclear homogeneous pattern and anti-dsDNA antibodies confirm de autoimmune disorders described in these patients. The cellular immunodeficiency with AHP is an expression of great clinical variability of this disease and the importance of early diagnosis.
Key words: inmunodeficiencies, Parry Romberg syndrome, Progressive Hemifacial Athrophy.
INTRODUCCIÓN
La Atrofia Hemifacial Progresiva , también conocida como Síndrome Parry Romberg es una enfermedad degenerativa extremadamente rara y pobremente comprendida, caracterizada por una lenta y progresiva atrofia facial unilateral que afecta al tejido celular subcutáneo, cartílago, tejido graso y estructuras óseas subyacentes, que frecuentemente se solapa con una condición conocida como esclerodermia lineal «en corte de sable». Su etiología es desconocida y el comienzo es lento, usualmente durante las dos primeras décadas de la vida, mas a menudo entre los 5 y 15 años de edad.1-4
Aunque esta enfermedad de aparición esporádica puede afectar ambos sexos, la incidencia es mayor en mujeres que en hombres, en una proporción aproximada de 3:2. La prevalencia es de al menos 1/700 000. La condición es observada en el lado izquierdo de la cara con mayor frecuencia que en el lado derecho, progresa durante varios años y luego se detiene.5,6
Esta patología fue descrita primero por Caleb Parry en 1825 y posteriormente desarrollada por Moritz Romberg y Eduard Henoch en 1846. Fue el neurólogo alemán Albert Eulenburg el primero en usar el término descriptivo de «Atrofia Hemifacial Progresiva» para esta enfermedad en 1871.5
Los hallazgos clínicos no se limitan solo a los cambios estructurales faciales, sino que pueden además presentarse manifestaciones neurológicas, oculares, cutáneas y en miembros y tronco, así como también algunas alteraciones dentarias. Dentro de las complicaciones neurológicas, la epilepsia focal es la manifestación cerebral más común y en algunos pacientes se asocia a un cuadro de hemiatrofia cerebral con convulsiones focales intratables y hallazgos histológicos en la biopsia de cerebro, indistinguibles de la encefalitis de Rasmussen. También puede aparecer migraña y dolor facial por neuralgia del trigémino, entre otras. Estas complicaciones pueden estar asociadas con anormalidades cerebrales del mismo lado de la atrofia facial, aunque raramente puede estar afectado el hemisferio cerebral contralateral.3,7-9 Las manifestaciones oculares más frecuentemente asociadas con el síndrome Parry Romberg incluyen el enoftalmo, retracción del párpado, ptosis palpebral, estrabismo restrictivo, disfunción del nervio motor ocular , síndrome de Horner, sensibilidad corneal reducida, queratopatía, episcleritis, uveítis, neuroretinitis, vasculitis retinal y disturbios pigmentarios de la retina.10-12
El diagnóstico de la Atrofia Hemifacial Progresiva puede realizarse solo sobre la base de la historia y el examen físico en un paciente que presente asimetría facial. En aquellos que además presenten síntomas neurológicos tales como migraña o convulsiones, la realización de un electroencefalograma y una Resonancia Magnética Nuclear (RMN) del cerebro constituyen la modalidad de elección. También en estos casos puede ser de utilidad la indicación de una punción lumbar y la determinación de autoanticuerpos en el suero del paciente.5,6
Debe realizarse el diagnóstico diferencial principalmente con el síndrome de Goldenhar, que es un desorden extraño que aparece al nacimiento, con las Lipodistrofias, la panatrofia de Gower y con la esclerodermia lineal en corte de sable, la cual incluye anormalidades oculares, orales y neurológicas similares al síndrome Parry Romberg.6,13
Hasta donde se conoce, no se ha reportado en la literatura la asociación de este síndrome a algún tipo de inmunodeficiencia, por lo que resulta de interés la presentación de este caso de atrofia hemifacial progresiva en el que se encontró una historia de infecciones recurrentes debido a un déficit en su sistema inmunológico.
PRESENTACIÓN DEL CASO
Paciente masculino, de la raza mestiza, de 5 años de edad, con antecedentes prenatales de feto cefálico, madre con anemia durante el último trimestre del embarazo, padre con ligera asimetría facial y abuela materna con enfermedad autoinmune (Tiroiditis y Diabetes Mellitus).
Nació por cesárea debido a desproporción cefalopélvica a las 40,3 semanas, con un apgar de 9/9, peso de 3860 gramos y una longitud supina de 51 cm. En las primeras 24 horas de nacido se le diagnosticó un ictero precoz que desapareció después de recibir tratamiento con fototerapia.
Su desarrollo psicomotor fue de características normales hasta la edad de 3 años en que apareció una zona de encanecimiento (piebaldismo) occipital derecha, con mancha hipocrómica de 0,5 cm en la piel subyacente, por lo que se sospechó que se trataba de un vitiligo. La biopsia realizada en dicha zona reveló como diagnóstico un Nevo de Bécquer. Recibió tratamiento con Melagenina durante varios meses.
A la edad de 4 años comenzó a presentar atrofia de la hemicara derecha incluida la oreja ipsilateral después de cuadro emético y febril, con progresión hasta los 5 años y fue este el motivo de remisión a las especialidades de Genética, Inmunología, Neurología y Maxilofacial.
En la primera consulta de Genética se encontró al examen físico una asimetría facial, dada por atrofia hemifacial derecha que involucraba piel y tejido celular subcutáneo, con zona de hiperpigmentación que rodeaba la oreja derecha. En región occipital del mismo lado se observó la presencia de una mancha hipocrómica con mechón de canas a ese nivel, así como ligera alopecia circunscrita a la región temporal del lado afectado, sin otras dismorfias importantes.
En Rayos X de cráneo realizado (vistas anteroposterior y lateral) no se apreciaron alteraciones óseas
A los 10 meses del inicio de la enfermedad, se evaluó nuevamente y se notó marcada progresión de la atrofia hemifacial derecha, con área de depresión que recuerda la estocada de sable, pérdida de la grasa y del tejido celular subcutáneo con visualización de la anatomía del arco cigomático, además de atrofia importante del lóbulo de la oreja de ese lado (oreja derecha 0,9 cm más pequeña que la izquierda) con plegamiento del hélix y desviación del mentón hacia el lado derecho. También se apreció un notable incremento de la pigmentación cutánea que abarcaba toda la hemicara afectada y aumento del área alopécica, sin variación ostensible en la zona de canicie (Figura).
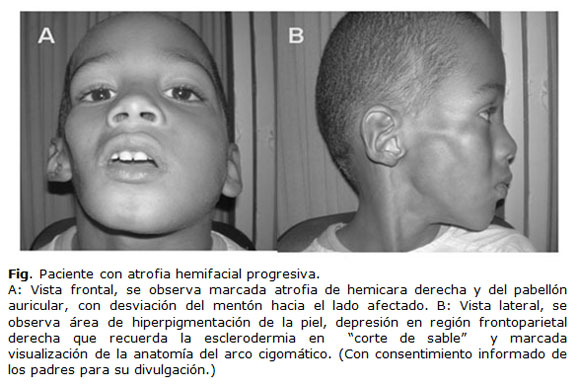
No se encontraron alteraciones dentarias en este paciente, manteniendo una oclusión funcional. La vista panorámica de los maxilares fue de resultado normal.
El examen oftalmológico realizado incluyó fondo de ojo, el cual reveló una ligera despigmentación de la retina del ojo derecho, con excavación de 0,5 mm y disminución ligera del grosor del iris asociada a zona de despigmentación.
Por la presencia de síntomas como irritabilidad, intranquilidad, tendencia a la depresión y llanto fácil ante una frustración el paciente fue valorado por la especialidad de Neurología, donde se le realizó estudio electroencefalográfico (EEG) que mostró como resultado un patrón epileptiforme, a pesar de la ausencia de convulsiones en este paciente y de tener examen neurológico normal.
El hemograma con diferencial mostró cifra de hemoglobina: 131 g/L (VN: 130-160 g/L), leucocitos totales normales: 14,5 x 109 /L (VN: 5 -14,5 x 109 /L) conteo absoluto de neutrófilos normal: 7,9 x 10 9/L (VN: 1,5 - 8 x 109 /L) normalidad de la cifra de linfocitos: 4,6 x 10 9/L (VN: 1,5 - 7 x 109 /L), con eosinófilos elevados 1,16 (VN: 0,05- 0,5 x 109 /L) y monocitos normales: 0,7 (VN: 0,15- 0,9 x 109 /L). El conteo de plaquetas fue de 240 x 109 /L (VN: 150-350 x 109 /L).
En los estudios inmunológicos realizados se obtuvieron concentraciones normales de las inmunoglobulinas (Ig) séricas IgG: 14,0 g/L (VN: 5,93 16,57 g/L), IgA: 1,92 g/L (VN: 0,5 2,30 g/L) e IgM: 0,84 g/L (VN: 0,29 1,20 g/L). La IgE mostró valores elevados de 172,1 UI/ml (VN ).Se encontraron valores normales de los componentes C3 y C4 del Sistema del Complemento. El valor de C3 fue de 1,28 g/L (VN: 0,9 1,70 g/L), mientras que el de C4 fue de 0,23 g/L (VN: 0,2 0,48 g/L). Los niveles de inmunocomplejos circulantes (ICC) fueron de 0,08 (VN: 0 0,130). La cuantificación de las subpoblaciones linfocitarias mostró una disminución en el número de linfocitos T CD2 positivos:59 % (VN:63 79 %) y de los CD4 positivos: 24 % (VN:40 65 %), con cifras incrementadas de las células T CD8 positivas: 40 % (VN:17 32 %) y valor normal del marcador de células B CD19: 28 % (VN:18 36 %).
La evaluación de la función fagocítica de los polimorfonucleares neutrófilos medida a diferentes intervalos de tiempo, mostró normalidad de los resultados obtenidos, con valor de 51,01 % a los 15 minutos (VN: 22,99 53,95 %) y de 17, 56 % a los 60 minutos (VN: 6,63 28,43 %).
Otras determinaciones realizadas fueron la Proteina C Reactiva (PCR) y el Factor Reumatoideo ( FR) cuyos resultados fueron negativos. En el estudio de autoanticuerpos se encontró una positividad de los anticuerpos antinucleares (ANA) con patrón homogéneo y de los anticuerpos anti- ADN de doble cadena (Anti ADN-dc).
La evaluación de los resultados del estudio inmunológico realizado llevó a la conclusión de que el paciente presentaba una inmunodeficiencia celular (déficit predominante de células T), por lo cual recibió atención y seguimiento periódico por consulta de Genética para la valoración sistemática de sus anomalías craneofaciales, así como también por las consultas de Inmunología, Neurología, Oftalmología y Máxilofacial.
Desde el punto de vista inmunológico se orientaron medidas de higiene personal y ambiental para impedir la recurrencia de los procesos infecciosos: evitar el contacto con personas enfermas, no exponerse a ambientes contaminados por gérmenes, suprimir la administración de vacunas que contengan gérmenes vivos atenuados y evitar, siempre que sea posible, el uso de procedimientos invasivos. Ante la presencia de infección se recomendó el tratamiento con antibióticos de amplio espectro según el resultado de los cultivos.
El tratamiento inmunológico consistió en la administración de Levamisol por vía oral en la dosis de 2,5 mg/Kg de peso 1 vez a la semana durante 8 semanas, seguido por el Factor de transferencia en dosis de una unidad semanal durante 8 semanas, por vía subcutánea o intramuscular. Posteriormente, ante la aparición de neutropenia relativa, se decidió suspender el Levamisol y continuar el tratamiento con factor de transferencia en la misma dosis indicada durante 16 semanas. Se le realizó después hemograma con diferencial evolutivo y se le indicó de nuevo tratamiento con Factor de transferencia semanal durante 16 semanas más.
DISCUSIÓN
La Atrofia Hemifacial Progresiva, ampliamente referida como Síndrome Parry-Romberg o enfermedad de Romberg (OMIM-141300), es un proceso patológico raro caracterizado por una atrofia adquirida, idiopática y limitada de un lado de la cara, que de forma variable involucra piel, tejido celular subcutáneo, grasa, músculos y menos frecuentemente las estructuras óseas subyacentes. Como su nombre sugiere esta enfermedad tiene una presentación unilateral, sin embargo, la manifestación bilateral se ha reportado en un 5-10 % de los casos. 14, 15.También se ha descrito en este síndrome atrofia del miembro inferior contralateral.16
Desde la primera descripción de la enfermedad en 1825, este síndrome ha suscitado numerosas interrogantes y reflexiones acerca de su fisiopatología, su expresión clínica variable y su progresión. 17 Entre los factores posibles involucrados en la patogénesis se encuentran los traumatismos, la herencia, infecciones virales, disturbios endocrinos y la autoinmunidad. 18 Aunque el mecanismo causal de la enfermedad es controvertido, la hipótesis autoinmunitaria se ha postulado como la más probable apoyada en la presencia de autoanticuerpos con un índice mayor que la población general, la superposición con escleroderma, vitiligo, uveitis y bandas oligoclonales en el líquido cefalorraquídeo. 6,7, 19 Investigaciones recientes han propuesto a la disfunción del sistema nervioso simpático como la teoría más aceptada para explicar la etiopatogenia de esta enfermedad. 16 En el caso que se presenta la etiología autoinmune es la que mejor explicaría la aparición del síndrome, por la detección de autoanticuerpos en el suero del paciente tales como ANA (patrón homogéneo) y Anti-ADN dc y tener antecedentes familiares de autoinmunidad. Estos hallazgos han sido comunicados por otros autores.20-23
Los signos clínicos que incluyen la hemiatrofia facial, la hiperpigmentación de la piel, la zona de canicie con mancha hipocrómica a ese nivel, el patrón eplileptiforme en el EEG y las alteraciones encontradas en el fondo de ojo, han coincidido con lo reportado en otros casos de la literatura revisada. 12, 18-21 Esta rara condición es más frecuente en hombres que en mujeres. Ren y colaboradores en su estudio clínico sobre Parry Romberg que se extendió de 1997 a Mayo de 2006, encontraron que de 35 pacientes afectados 24 eran del sexo femenino y solo 11 del masculino. 24 El caso que aquí presentamos es varón, lo cual concuerda con algunos autores.21-23
En la atrofia facial generalmente está involucrada la hemicara izquierda. 20,22,23 Sin embargo, en el caso que aquí se presenta la hemiatrofia facial fue derecha, lo cual ha sido descrito con anterioridad en otros enfermos con este síndrome.6,21,25,26
Además de las manifestaciones propias del síndrome Parry Romberg al paciente se le diagnosticó una inmunodeficiencia celular (déficit predominante de células T), lo cual no ha sido comunicado en la literatura revisada para esta entidad. En el estudio de las subpoblaciones linfocitarias se encontró una disminución en los valores de los marcadores linfocitarios T CD2 y CD4. La inmunidad celular, que forma parte de la respuesta inmunitaria adaptativa, es la función efectora de los linfocitos T y actúa como mecanismo de defensa frente a los microorganismos que sobreviven en el interior de los fagocitos o que infectan células no fagocíticas. Después del reconocimiento antigénico a través de receptores específicos, los linfocitos T experimentan un proceso de activación que conlleva a su proliferación, expansión clonal, formación de células de memoria, secreción de citocinas y/o respuesta citotóxica. La activación efectiva de los linfocitos requiere de señales coestimuladoras que contribuyan con la señal generada a través del receptor antigénico, entre las que juega un papel primordial la molécula CD28. 27-29 Las infecciones virales y bacterianas que ha padecido el paciente podrían explicarse por la disminución que presenta de las subpoblaciones linfocitarias T. El incremento encontrado en el valor del marcador de células T CD8 podría obedecer a un mecanismo inmunológico compensatorio por la marcada disminución de la subpoblación auxiliadora inductora de la respuesta inmune (CD4+).
El hallazgo de una inmunodeficiencia celular en un paciente con Atrofia hemifacial progresiva es expresión de la gran variabilidad clínica de esta rara enfermedad y de la importancia que adquiere un diagnóstico temprano de la misma. Se hace necesario un adecuado plan de tratamiento, que debe ser ejecutado por un equipo multidisciplinario de especialistas, para lograr un buen manejo funcional y psicológico del paciente con la finalidad de mejorar su calidad de vida.
REFERENCIAS BIBLIOGRÁFICAS
1. Grimaldi M, Gentile P, Labardi L, Silvi E, Trimarco A, Cervelli V. Lipostructure technique in Romberg syndrome. J Craniofac Surg 2008 Jul;19(4):1089-91.
2. Kacinski M, Biedron A, Zajac A, Steczkowska M. Diagnostic difficulties of paroxysmal symptoms in a boy with Parry Romberg syndrome. Neurol Neurochir Pol 2010 May-Jun;44(3):297-303.
3. Maletic J, Tsirka V, Ioannides P, Karacostas D, Taskos N. Parry-Romberg Síndrome with Localizad Scleroderma. Case Rep Neurol 2010 Jun; 2(2):57-62.
4. WójcickiP, Zachara M. Surgical treatment of patients with Parry Romberg syndrome. Ann Plast Surg 2011 Mar;66(3):267-72.
5. Wikipedia. Parry-romberg syndrome. Disponible en: http//pillen.wikipedia.wikipedia.org/wiki/Parry%E2%80%93Romberg-syndrome (Visitada 18-8-11, actualizada 22-9-11)
6. Stone J. Parry Romberg síndrome. Practical Neurology 2006 August 22; 6(3):185-8.
7. Buonanotte F. Síndrome de Parry Romberg: variaciones clínicas y consideraciones neurológicas. Rev Neurol 2007 Jul; 45(3):189-90.
8. Kakisaka Y, So NK, Jones S, Wang ZI, Mosher JC, Alexopoulos AV et al. Intractable focal epilepsy contralateral to the side of facial atrophy in Parry-Romberg syndrome. Neurol Sci 2011 Feb;33(1):165-8. DOI 10.1007/s10072-11-0643-z.
9. Viana M, Glastonbury CM, Sprenger T, Goadsby PJ. Trigeminal neuropathic pain in a patient with progressive facial hemiatrophy (parry-romberg syndrome). Arch Neurol 2011 Jul;68(7):938-43.
10. Nasser O, Greiner K, Amer R. Unilateral optic atrophy preceding Coats Disease in a girl with Parry-Romberg syndrome. Eur J Ophthalmol 2010 Jan-Feb;20(1):221-3.
11. Zubcov-Iwantscheff AA, Thomke F, Goebel HH, Bacharach-BuhlesM, Cordey A, Constantinescu CS et al. Eye movement involvement in Parry-Romberg Syndrome: a clinicopathologic case report. Strabismus 2008 Jul-Sep;16(3):119-21.
12. Dawczynski J, Thorwarth M, Koenigsdoerffer E, Schultze-Mosgau S. Interdisciplinary treatment and ophthalmological findings in Parry-Romberg syndrome. J Craniofac Surg 2006 Nov;17(6):1175-6.
13. Chiang KL, Chang KP, ong TT, Hsu TR. Linear scleroderma «en coup de sabre»: inicial presentation as intractable partial seizures in a child. Pediatr Neonatol 2009 Dec;50(6):294-8.
14. Vaienti L, soresina M, Menozzi A. Parascapular free flat and fat grafts: Combined surgical methods in morphological restoration of hemifacial Progressive atrophy. Plast Reconst Surg 2005 Sept;116(3):699-711.
15. Ye XD, Li CY, Wang C, Yu YS. Superficial temporal fascial flap plus lipofilling for facial contour reconstruction in bilateral progressive facial hemiatrophy. Aesthetic Plast Sur 2010 Aug;34(4):537-7.
16. Duymaz A, Karabekmez FE, Keskin M, Tosun Z. Parry-Romberg syndrome: facial atrophy and its relationship with other regions of the body. Ann Plast Surg 2009 Oct;63(4):457-61.
17. Ruhin B, Bennaceur S, Vercke F, Louafi S, Seddiki B, Ferri J. Progressive hemifacial atrophy in the young patient: phisiopathologic hypotheses, diagnosis and therapy. Rev Stomatol Chir Maxillofac 2010 Dec;101(6):287-97.
18. Pinheiro TP, Silva CC, Silveira CS, Botelho PC, Pinheiro MG, Pinheiro Jde J. Progressive Hemifacial Atrophycase report. Med Oral Patol Oral Cir Bucal 2006 March 1;11(2):E112-4.
19. Okumura A, Ikuta T, Tsuji T, Fujatsu H, Naganawa S, Kato K, et al. Parry-Romberg syndrome with a clinically silent white matter lesion. AJNR Am J Neuroradiol 2006 Sept;27(8):1729-31.
20. Thapa R, Ghosh A, Dhar S. Parry-Romberg Syndrome with Band-like Alopecia. Indian J Pediat 2009 Jul;76(s/n):760.
21. Gulati S, Jain V, Garg G. Parry Romberg Syndrome. Indian J Pediat 2006 May;73(s/n):448-9.
22. Galarza C, Gutiérrez E, Ramos W, Macetas R, Mendoza M, Gómez A, et al. Síndrome de Parry Romberg. Dermatol Peru 2006;16(2):151-4.
23. Mizraji G, Yacovino D, Sinay V, De simone V, Kadar J, Chade A, et al. Síndrome de Parry Romberg. Rev Neurol Argentina 2001 Nov; 26:78-82.
24. Ren M, Teng L, Gui L. Clinical study on correction of hemifacial atrophy with free anterolateral thigh adipofascial flap. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi 2007 Sop;21(9):913-6.
25. Kumar AA, Kumar RA, Shantha GP, Aloogopinathan G. Progressive hemifacial atrophy Parry Romberg syndrome presenting as severe facial pain in a young man: a case report. Cases J 2009 Jul 2;6776.
26. Lane T, Cheung J, Schaffer J. Parry-Romberg syndrome with coexistent morphea. Dermatology Online Journal 2008 Oct;14(10):21-22.
27. Abbas K, Lichtman A, Pober J. Mecanismos efectores de la inmunidad Celular. En: Inmunología celular y molecular.4 ed. Madrid. McGraw-Hill. Interamericana; 2002.p.303-21.
28. Marsán V, Villaescusa R, del Valle LO, Arce A, Torres I, Macías C. Inmunodeficiencia primaria combinada: Presentación de un caso. Rev Cubana Hematol Inmunol Hemoter 2001 ene-abr;17(1):55-8.
29. Barboza L, Martens ML, Salmen S, Peterson DL, Montes H, Berruela L. Déficit en señales de activación y coestimulación en linfocitos T CD4+ de pacientes infectados con el virus de la hepatitis C. Invest Clin 2008 Feb;49(3):353-67.
Recibido: 1 de noviembre de 2013.
Aprobado: 17 de enero de 2013.
Dra. Miriam de la C. Sánchez Segura. Instituto de Hematología e Inmunología. La Habana, Cuba. E mail: ihi@infomed.sld.cu

