










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Investigaciones Biomédicas
versión On-line ISSN 1561-3011
Rev Cubana Invest Bioméd v.16 n.2 Ciudad de la Habana jul.-dic. 1997
Trabajos de Revisión
Instituto Superior de Ciencias Médicas de La Habana Centro de Investigaciones BiomédicasBases moleculares de la hipertrofia ventricular izquierda. papel del estrés oxidativo
Dr. Félix Broche Valle, Lic. Marisol Peña Sánchez, Dra. Ela M. Céspedes Miranda, Dr. José C. García Piñeiro y Dr. José Castillo HerreraRESUMEN
La hipertensión arterial (HTA) como enfermedad primaria es una de las primeras causas de morbilidad y mortalidad general en muchos países. La cardiopatía isquémica y otras formas clínicas de la enfermedad cardiovascular aterosclerótica se presentan con mayor frecuencia en los pacientes hipertensos. La aparición y severidad de las manifestaciones de isquemia miocárdica se incrementan si coexiste hipertrofia ventricular izquierda. En el miocardio hipertrófico se producen eventos de isquemia/reperfusión durante cada ciclo cardíaco. En estas condiciones, diversos factores conducen al estrés oxidativo cuyas consecuencias se expresan por alteraciones morfofuncionales en el corazón y otros sistemas. En este trabajo se presentan los principales mecanismos conocidos que aportan las bases moleculares de la hipertrofia ventricular y de la pared arterial en los pacientes hipertensos, así como el origen y significado fisiopatológico del estrés oxidativo en esta entidad con el propósito de contribuir a la sustentación teórica de los criterios para diagnóstico, pronóstico, evolución y tratamiento del paciente hipertenso con hipertrofia ventricular izquierda.Descriptores DeCS: HIPERTROFIA VENTRICULAR IZQUIERDA/fisiopatología; ESTRES OXIDATIVO; SISTEMA RENINA-ANGIOTENSINA; HIPERTENSION/complicaciones.
FISIOPATOLOGÍA DE LA HIPERTROFIA VENTRICULAR
Las enfermedades cardiovasculares, y en particular la hipertensión arterial (HTA) se ubican entre las 3 primeras causas de morbilidad y mortalidad general en los países desarrollados. Entre los factores implicados en la fisiopatología de la hipertensión arterial se encuentran:
- Reflejos barorreceptor y quimiorreceptor.
- Isquemia del sistema nervioso central.
- Sistema renina angiotensina.
- Otras sustancias reguladoras del tono vascular con actividad paracrina o endocrina (por elemplo: endotelinas, óxido nítrico, prostanoides, quininas, histamina, etcétera).
- Actividad del sistema nervioso vegetativo.
- Excreción renal de Na y agua.
- Distensión de las paredes vasculares.
- Intercambio de líquido entre los compartimientos intra y extravascular.
El estudio de Framingham3,4 demostró que la HTA es capaz de conducir a episodios de isquemia miocárdica en individuos con predisposición genética, fenómeno que se exacerba si coexiste hipertrofia ventricular izquierda (HVI).5
El propio estudio reportó que la existencia del HVI aumentaba hasta 10 veces la mortalidad por enfermedad cardiovascular lo que le confiere a ésta el carácter de factor de riesgo independiente.6
El progresivo crecimiento de la pared ventricular se interpreta como respuesta compensatoria tendente a reducir el estrés parietal sistólico sin modificar el radio de la cavidad cardíaca ante el incremento de la resistencia periférica total.
La HVI no es una complicación tardía e irreversible de la HTA sino un fenómeno temprano y modificable en la evolución de la enfermedad.6 Varios estudios sugieren que la capacidad para desarrollar HVI en los pacientes hipertensos está genéticamente determinada.5
Todos los modelos experimentales coinciden en que no aparecen señales de mitosis en corazones adultos hipertrofiados; en consecuencia, el crecimiento cardíaco se realiza únicamente a través del aumento en la síntesis de las proteínas contráctiles y de la matriz extracelular (MEC)7 y la hipertrofia de los miocitos existentes.8
La MEC es un complejo sistema constituido básicamente por colágeno y fibronectina. También aparecen laminina, elastina, otras glicoproteínas, proteoglicanos y ácido hialurónico.7
Los colágenos tipos I y III de colágenos son los más abundantes y se comportan como material de relleno en el proceso de reparación posnecrótica del miocardio.7 El colágeno tipo IV aparece en forma de filamentos cercanos a la lámina basal de los miocitos.9
En el miocardio hipertrófico se produce un remodelaje de la arquitectura tisular con predominio de bandas fibrosas de alto contenido de colágeno tipo I; en este entorno se dificulta la propagación del impulso cardíaco y la difusión de los nutrientes, en particular el oxígeno.7
El factor transformante B1 (TGF B1) se ha señalado como el principal regulador transcripcional de la síntesis de colágeno y fibronectina en el miocardio.10 Es un factor de crecimiento multifuncional de naturaleza polipeptídica, cuyos receptores de membrana se han identificado hasta el presente en todos los tipos de células estudiadas. Los factores TGF B1, B2 y B3 parecen ser funcionalmente equivalentes.11
La naturaleza de la respuesta depende del tipo celular, las condiciones de crecimiento y diferenciación, así como de la presencia de otros factores de crecimiento.11 Se considera además que inhibe la degradación de la MEC. Sus efectos sobre el crecimiento y la diferenciación son variados.11 Igualmente se reconoce su papel en la proliferación fibroblástica del miocardio y el músculo liso vascular.12,13
La actividad del TGF B está sujeta a mecanismos de regulación endocrina (mediada por la norepinefrina y angiotensina II) y autocrina.12,13
La participación del Ca2+ como mensajero intracelular está asociada al sistema de la fosfolipasa C (PLC) cuya actividad es promovida por proteínas GTP-dependientes (Proteína G).6
La PLC genera inositol trifosfato (IP3) y diacilglicerol (DAG). El IP3 se comporta como segundo mensajero soluble en el citosol y promueve la apertura de los canales de calcio en las membranas citoplasmática y del retículo endoplásmico. Esta respuesta rápida de aumento del calcio intracelular activa varios sistemas dependientes de la proteína calmodulina.6
El DAG permanece en la membrana citoplasmática formando un complejo con la proteína quinasa C (PKC) sensible al Ca2+. El complejo PKC-DAG-Ca2+ inicia una cascada de fosforilación de proteínas reguladoras intracelulares cuyo resultado incluye el incremento de la síntesis de colágeno y miosina entre otros efectos.6
En la membrana citoplasmática también se localiza un intercambio Na+-H+ que bombea H+ activamente hacia el espacio extracelular y cuya actividad se incrementa notablemente al ser fosforilado por el complejo PKC-DAG-Ca2+. La alcalinización del citosol se reconoce como factor activador transcripcional.6
Diferentes agonistas inducen el incremento del colágneo intra y extracelular a través de estos mecanismos u otros que en algunos casos se conocen sólo parcialmente. En la literatura se citan los factores de crecimiento derivados del endotelio (FCDEN) y plaquetas (FCDP), angiotensina II y las endotelinas.6
Las endotelinas son una familia de polipéptidos sintetizados por las células endoteliales y del músculo liso. Se les atribuye participación en la fisiopatología de la HTA por su efecto vasoconstrictor mediado por el aumento de la permeabilidad celular al Ca2+ y activación de la PKC.14-17
Con diferentes líneas celulares se ha reportado incremento en la síntesis de factores de crecimiento, colágeno y fibronectina inducidos por estas moléculas (Moreland S. Bristol-Myers Squibb, Pharmaceutical Research Institute, 1955. Comunicación personal).
Se ha sugerido el papel estimulador del Ca2+ y un mecanismo de inducción autocrina de la síntesis de endotelinas.14-15
No todos los antihipertensivos reducen la HVI a pesar de garantizar un adecuado control de las cifras tensionales. Los diuréticos y vasodilatadores del tipo de la hidralacina no modifican la masa ventricular. Por otra parte, la metildopa reduce la HVI sin relación alguna con las cifras de tensión arterial. Entre ambos extremos los inhibidores de la enzima convertidora de angiotensina I (IECA), los antagonistas del Ca2+, las dihidropiridinas, el prazosín, la clonidina y los beta bloqueadores sin actividad simpaticomimética intrínseca inducen paralelamente la reducción de las cifras tensionales y la regresión de la HVI.6
PAPEL DEL SISTEMA RENINA-ANGIOTENSINA
El octapéptido angiotensina II es el agente vasoconstrictor endógeno más potente que se conoce.18Su formación es el resultado de una cascada de reacciones proteolíticas que se inicia por la renina, proteasa sintetizada en el aparato yuxtaglomerular del riñón, al actuar sobre una globulina del plasma y liberar el decapéptido angiotensina I; posteriormente la enzima convertidora de angiotensina I (IECA) separa 2 aminoácidos para formar la angiotensina II.18
El sistema renina-angiotensina interviene en el control a corto y largo plazos de la presión arterial media a través del efecto presor directo de la angiotensina II (mediado por la reducción de la concentración intracelular de monofosfato cíclico de guanosina -GMPc-) y la retención hidrosalina promovida por la aldosterona (cuya secreción es estimulada por la angiotensina II).19-21
La conversión de angiotensina I en II puede ser catalizada también por una enzima convertasa específica del miocardio.22
Del receptor de angiotensina II (ATr) se conocen 2 subtipos.23 El subtipo AT1 es un receptor acoplado a la Proteína G que induce la disminución de la concentración intracelular de GMPc por estimulación de enzimas fosfodiesterasas, mecanismo que activa la contracción muscular. Igualmente promueve la fosforilación de residuos de tirosina.24 En el miocardio sólo aparece este subtipo de receptor.6
Por otra parte, el subtipo AT2 actúa por inhibición directa de la enzima guanilato ciclasa y activa enzimas fosfotirosina fosfatasas.24
Actualmente se acepta la asociación entre eventos de fosforilación-desfosforilación de residuos de tirosina y los procesos de crecimiento y diferenciación celular.25
Los efectos de la angiotensina II también se vinculan a la liberación de aldosterona y norepinefrina.6
Desde su introducción en los años 80, los IECA se han utilizado en el tratamiento de la HTA esencial y la insuficiencia cardíaca congestiva asociada a infarto agudo del miocardio26 y HTA.22,27-31
Se ha reportado que el enalapril incrementa el número de mitocondrias en los cardiomiocitos.32 También se le atribuye un efecto protector miocárdico durante y después de un infarto agudo del miocardio.31 Otros estudios sugieren que reduce la insulinorresistencia en los pacientes hipertensos.33
Estudios recientes en ratones tratados con enalapril durante 4 a 11 semanas demostraron un incremento de la defensa antioxidante plasmática mediada por las enzimas superóxido dismutasa (Cu-Zn y Mn SOD) y glutatión peroxidasa (GPx).34
ESTRÉS OXIDATIVO EN CONDICIONES DE ISQUEMIA/REPERFUSIÓN. SU PAPEL EN LA FISIOPATOLOGÍA DE LA HVI
La difusión anormal del oxígeno en el miocardio hipertrófico genera un déficit energético progresivo en tanto se vincula también a los mecanismos moleculares del estrés oxidativo en condiciones de isquemia/reperfusión.35Se sabe que en el corazón humano hipertrofiado el número de capilares miocárdicos no aumenta proporcionalmente con el aumento de masa.36 En el miocardio hipertrófico se reduce adicionalmente el flujo sanguíneo durante la sístole cardiaca, en particular a nivel del tejido subendocárdico.
En consecuencia, el tejido está sometido a eventos cíclicos de isquemia/reperfusión debido al aumento de la distancia capilares-miocitos, la disminución de la relación número de capilares/masa muscular36 y la reducción marcada del flujo coronario en la fase sistólica (figura).
Como expresión de la "paradoja del oxígeno", entre el 2 y el 5 % del oxígeno en los organismos aerobios es reducido parcialmente con formación de "radicales libres" como el anión superóxido y el radical hidroxilo.7 Estas y otras moléculas como oxígeno singlete y peróxido de hidrógeno (H2O2) en su conjunto se identifican como especies reactivas del oxígeno (ERO).
Una vez generadas, las ERO reaccionan rápidamente en posiciones susceptibles a la oxidación, virtualmente con cualquier biomolécula accesible a nivel de su sitio de formación.
El organismo dispone de mecanismos de defensa antioxidante8,37,38 que incluyen:
- Barreras fisiológicas que confronta el oxígeno a su paso desde el aire inspirado hasta las células.
- Enzimas como SODs, catalasa (CAT), GPx, glutatión reductasa (GRd) y sistemas regeneradores de NADPH H+.
- Adecuados niveles tisulares y plasmáticos de compuestos antioxidantes directos como las vitaminas A, E y C, glutatión reducido (GSH) y coenzima Q.10
- Transportadores de metales de transición como la transferrina y la ceruloplasmina.
En la literatura se reportan algunas evidencias que sugieren la participación del estrés oxidativo en la fisiopatología de la HTA y sus complicaciones.
Varios autores han señalado la disminución de los niveles de SOD y GSH asociados a los cambios vasculares en la HTA41 así como el aumento de la actividad de xantana oxidasa (XO) en el corazón de ratas hipertensas, con disminución de la relación XD/XO.42
El "consumo" de NO* en presencia de altas concentraciones de anión superóxido se ha sugerido como elemento inductor de vasoconstricción asociado también al daño de las células endoteliales y del músculo liso vascular por el radical peroxinitrito.43
Actualmente se acepta la teoría del estrés oxidativo en condiciones de isquemia-reperfusión teniendo en cuenta el papel reconocido de las ERO en la generación de daño celular.
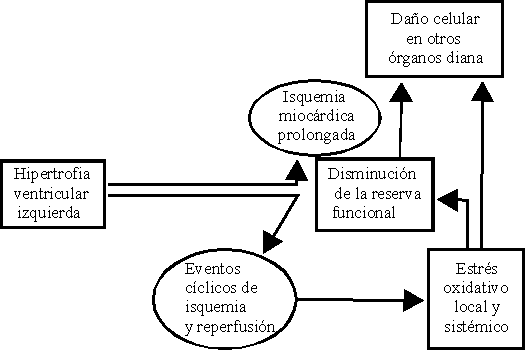
La isquemia es fuente de daño y muerte celular a partir del agotamiento de las reservas intracelulares de ATP. La incapacidad para el mantenimiento del balance iónico, las modificaciones en la homeostasis del calcio,44 la disfunción mitocondrial, la activación de enzimas como la fosfolipasa A2 y otras vinculadas al metabolismo del ácido araquidónico, son algunos de los eventos iniciales del daño celular por isquemia/reperfusión.6
Adicionalmente aparecen modificaciones en la barrera endotelial, activación leucocitaria y coagulación intravascular;6 pero la reperfusión, factor esencial en la recuperación del tejido isquémico, se acompaña paradójicamente de efectos de daño celular45 que pueden focalizarse a nivel de:
- Generación de ERO y activación de enzimas Ca2+ dependientes.
- Acumulación de leucocitos y plaquetas con coagulación intravascular y edema intersticial que comprometen la perfusión.
- Respuesta inflamatoria aguda mediada por los neutrófilos y macrófagos tisulares.
La XO en la mayoría de los tejidos existe en su forma deshidrogenasa (XD) (no generadora de ERO) que puede convertirse en la forma XO por proteólisis parcial entre otros factores. A su vez se reconoce su papel en la generación de radical superóxido por los leucocitos activados.48
Las células fagocíticas liberan radical superóxido que se transforma en radical hidroxilo y H2O2, este último actúa como intermediario en la formación de ácido hipocloroso catalizada por la enzima lisosomal mieloperoxidasa. La citotoxicidad del H2O2 y el ácido hipocloroso se ha verificado en múltiples tejidos.6,47
La velocidad de generación del radical superóxido se incrementa al aumentar la concentración intramitocondrial de oxígeno o reducirse la intensidad del transporte electrónico,47 condiciones también asociadas a los eventos de isquemia/reperfusión.
La reoxigenación de inicio se asocia al incremento en la formación de radical superóxido a causa de la baja disponibilidad de adenosín difosfato (ADP) por degradación de los nucleótidos de adenosina durante la fase isquémica que junto a otros factores se imponen como limitantes para la fosforilación oxidativa.48
Las interleuquinas 1 y 2, el factor de necrosis tumoral (TNF) y otras son capaces de inducir la activación de la isoenzima óxido nítrico sintetasa (no dependiente del calcio) a nivel de neutrófilos y macrófagos, evento asociado al proceso inflamatorio en varios órganos y en el que se le atribuye al aumento en la producción de NO* un papel central con relación al incremento de la permeabilidad vascular y otras alteraciones de la homeostasis local.49
Varios trabajos han demostrado que la defensa antioxidante es variable en las diferentes capas de la pared miocárdica. En el tejido subendocárdico se detecta menor actividad de catalasa y concentraciones más bajas de vitamina E y glutatión reducido asociados con un mayor índice de peroxidación lipídica respecto al resto del miocardio, en tanto la actividad de las enzimas antioxidantes parece ser uniforme en todo el tejido transmural.50
Ji LL y colaboradores51 detectaron que, en animales de experimentación sometidos a isquemia/reperfusión, disminuyó el contenido total de GSH y la actividad de GRd y GPx, así como un incremento de la peroxidación lipídica con respecto a los animales controles. La inducción previa de hipertrofia ventricular mejoró la resistencia del órgano al daño por isquemia/reperfusión, probablemente por un efecto de precondicionamiento.
En el origen del precondicionamiento isquémico se considera entre otros mecanismos, la inducción de la transcripción de los genes del "shock térmico" promovida por las ERO y la depleción de ATP. Algunas proteínas del "shock térmico" poseen actividad de "chaperonas moleculares" favoreciendo las conformaciones nativas de ciertas proteínas claves en el metabolismo intermediario, lo cual explicaría sus efectos citoprotectores durante exposiciones sucesivas a situaciones de estrés a partir de la exposición al evento o la situación precondicionante.52,53
En condiciones de isquemia/reperfusión se manifiestan otros mecanimos de daño celular. Así, por ejemplo, se conoce el papel de las ERO en los mecanismos de apoptosis y el efecto aterogénico de la oxidación de las lipoproteínas de baja densidad (LDL).54
En la literatura se aprecia evidente consenso en torno al efecto cardioprotector de diferentes combinaciones de antioxidantes. Recientemente, por ejemplo, viene comercializándose el carvedilol, molécula con efectos vasodilatador y antiproliferativo y capacidad antioxidante directa sobre radicales hidroxilo y peroxilo,57 cuyas perspectivas resultan alentadoras para la profilaxis y el tratamiento de la HVI.
CONCLUSIONES
La consideración de la HVI no sólo como consecuencia sino también como factor de riesgo independiente para las complicaciones cardíacas de la HTA, supone que los mecanismos moleculares que le dan origen sean objeto de investigaciones multidisciplinarias que posibiliten ampliar los conocimientos básico-clínicos y aporten las bases para nuevas estrategias de intervención con carácter profiláctico o terapéutico.En este sentido, la participación del estrés oxidativo como mecanismo de daño celular sistémico asociado a los eventos cíclicos de isquemia/reperfusión que se producen en el miocardio y la pared arterial hipertróficos constituye un factor patogénico adicional que también debe ser considerado en el perfeccionamiento de las estrategias de diagnóstico y tratamiento de la HTA y sus complicaciones.
SUMMARY
- Arterial hypertension (AHT) as a primary disease is one of the first causes of morbidity and general mortality in many countries. Ischemic cardiopathy and other clinical forms of the atherosclerotic cardiovascular disease are more common among hypertensives. The appearance and severity of the manifestations of myocardial infarction increase if left ventricular hypertrophy coexists. In the hypertrophic myocardium, events of ischemia/reperfusion take place during each cardiac cycle. Under these conditions, different factors lead to the oxidative stress, whose consequences are expressed by morph ofunctional alterations of the heart and other systems. In this paper, the main mechanisms giving molecular bases of ventricular hypertrophy ahod of the arterial wall in hypertensives, as well as the origin and physiopathologic significance of the oxidative stress in this disease, are presented aimed at contributing to the theoretical support of the criteria for diagnosis, prognosis, evolution and treatment of the hypertensive patient suffering from left ventricular hypertrophy.
Subject headings: HYPERTROPHY; LEFT VENTRICULAR/physiopathology; OXIDATIVE STRESS; RENIN-ANGIOTENSIN SYSTEM; HYPERTENSION/complications.
REFERENCIAS BIBLIOGRÁFICAS
- Stein J. Medicina interna. La Habana: Editorial Científico-Técnica, 1987,vol1,t1:573-90.
- Rigol RO, Pérez CF, Perea CJ, Fernández SJ, Fernández MJ. Medicina general integral. La Habana: Editorial Pueblo y Educación, 1987,t3:11-23.
- Chobaninan AV, Brecher PI, Haudenschild CC. Effects of hypertension and antihypertensive therapy on atherosclerosis: state of the art lecture. Hypertension 1986;8(Suppl 1):15-21.
- Kannell WB. Some lessons in cardiovascular epidemiology from Framingham. Am J Cardiol 1976;37:269--82.
- Kannell WB, Castelli WP, Mellumara AM, McKee PH, Fernleib M. Role of blood pressure in the development of congestive heart failure. N Engl J Med 1972;297:781-7.
- Coca A, Sierra A de la. Mecanismos patogenéticos de la hipertrodia cardíaca en la hipertensión arterial. Med Clin (Barc) 1991; 97:667-76.
- Pelouch V, Dixon I, Golfman L, Beamish R, Dhalla N. Role of extracellular matrix proteins in heart function. Mol Cell Biochem 1994;129:101-120.
- Page E, McCalister LP. Quantitative electron microscopic description of heart muscle cells. Application to normal, hyperthrophied and thyroxinstimulated heart. Am J Cardiol 1973;31:172-81.
- Bashey RI, Martínez-Hernández A, Jiminez SA. Isolation, characterization and localization of cardiac collagen type VI. Associations with other extracellular matrix components. Circ Res 1992;70:1006-1007.
- Eghbali M, Tomek R, Suthatme VP, Wood Ch, Bhambi B. Different effects of transforming growth factors and phorbol myristate acetate and cardiac fibroblasts. Regulation of fibrillar collagen mRNAs and expression of early transcription factors. Circ Res 1991;69:483-90.
- Sporm MB, Roberts AB. The transforming growth factors Bs. En: Sporm MB, Roberts AB. Peptide growth factors and their receptors. 1990:
- Gibbons G, Pratt RE, Dzau VJ. Vascular smooth muscle cells hypertrophy vs hyperplasia. Autocrine transforming growth factor B1 expression determines growth response to angiotensin II. J Clin Invest 1992;90:456-61.
- Stouffer GA, Owens GK. Angiotensin II induced mitogenesis of spontaneously hypertensive rat derived cultured smooth muscle cells is dependent on autocrine production of transforming growth factor B. Circ Res 1992;70:820-8.
- Graier FW, Sturek M, Kukowets W. Ca2+ regulation and endothelial vascular function. Endothelium 1994;1:223-36.
- Lewis M, Shah A. Endothelial modulation of myocardial contraction. Endothelium 1994;1:237-43.
- Battistini B, O'Donnell LJ, Warner TD, Fournier A, Farthing MJ, Vane JR. Characterization of endothelin receptors in the isolated gall blader of the guinea pig evidence for an additional ET receptor subtype. Br J Pharmacol 1994;112(4):1244-50.
- Corder R, Vane JR. Radioimmunoassay evidence that the pressor effect of big endothelin-1 is due to local conversion to endothelin-1. Biochem Pharmacol 1995;49(3):375-80.
- Black D, Jones NF. Enfermedades renales. La Habana: EditorialE Científico-Técnica, 1985;t1:62.
- Hipertensión esencial. Clin Méd Norteam 1987.
- Lloyd HS, Samuel OT. Fisiopatología: principios biológicos de la enfermedad. La Habana: Editorial Científico-Técnica, 1986;t2:893.
- Morgan R, Stewart JM, Vavrek RJ, Hassid A. Effects of bradykinin and angiotensin II on intracellular calcium dynamics in endothelial cells. Am J Physiol 1987;253:C588-98.
- Brechler V, Levens N, De Gasparo M, Bottari S. Angiotensin AT2 receptor mediated inhibition of particulate guanylate cyclase: a link with protein tyrosine phosphatase stimulation? Recept Chann 1994;2(2):79-87.
- Brechler V, Reichlin S, De Gasparo M, Bottari S. Angiotensin II stimulates protein tyrosine phosphatase activity through a G protein independent mechanism. Recept Chann 1994;2(2):89-97.
- Zeiher AM, Schchinger V. Coronary endothelial vasodilator dysfunction: clinical relevance and therapeutic implications. Z Kardiol 1994;83(Suppl 4):7-14.
- Geer P van der, Hunter T, Lindberg R. Receptor protein-tyrosine kinases and their signal transduction pathways. Annu Rev Cell Biol 1994;10:251-337.
- Secchi J. Effects of subchronic treatment with trandolapril and enalapril on cardiovascular morphologic alterations in the aged spontaneously hypertensive rat with heart failure. J Cardiovasc Pharmacol 1994;23(Suppl 4s):30-7.
- Kuchansk J. Significance of mitochondrial Ca2+ transport in ischemic injury and myocardial protection. Bratisl Lex Listy 1994;95(9):391-4.
- Vademecum internacional. 36 ed. Madrid: Medicom, 1995.
- Haznedaroglu IC. Effects of ACE inhibition on left ventricular dimensions and haemodynamics in systemic hypertension, a radionuclide and echocardiographic study. Cent Afr J Med 1995;41(4):118-23.
- Dahlof B, Pennert R, Hansson L. Reversal of left ventricular hypertrophy in hypertensive patients. A metaanalysis of 109 treatment studies. Am J Hypertens 1992;5:95-110.
- Enstrm J, Thulin T, Lindholm LH. Comparison between enalapril and lisinopril in mild-moderate hypertension. A comprehensive model for evaluation of drug efficacy. Blood Press 1992;1(2):102-7.
- Rengo F. Comparison of the safety and efficacy of dilapril with enalapril in patients with congestive heart failure. Am J Cardiol 1995;75(16):25F-8F.
- Vuorinen MH, Yki JH. Effects of enalapril on insulin resistance. Metabolism 1995;44(1):85-9.
- Cavanagh EM de. Oxidative stress and renal dysfunction in rats treated with enalapril. FEBS Lett 1995;36(1):22-4.
- Why HJ, Ansell H, Patel VB, Wassif WS, Reeves J, Sidding T, et al. Antioxidant status in hypertension and effects of angiotensin converting enzyme inhibition. Biochem Soc Trans 1995;23(2):224S.
- Romero D, Villalba Martin M, Bueno J. Papel de la prooxidación y la antioxidación en la etiología de la hipertensión arterial. Med Clin (Barc) 1991;97:542-51.
- Sohal RS. Superoxide anion radical production in different animal species. Mech Ageing Dev 1989;49:129--35.
- Chang LY, Molecular immunocytochemistry of the CuZn superoxide dismutase in rat hepatocytes. J Cell Biol 1988;107:2169.
- Pryor WA. A festschfift volume celebrating the 20th anniversary of superoxide dismutase. Free Rad Biol Med 1988;5:271.
- Broche VF, Romero SA, Olembe SE, Fernández E, Céspedes ME, García J, et al. Aprotinin effects related to oxidative stress in cardiosurgery with mechanical cardiorespiratory support. Ann NY Acad Sci 1996;793:521-4.
- Sagar S. Oxigen free radicals in essential hypertension. Roll Gell Brochen 1992;111(1-2):103-8.
- Kooij A. A reevaluation of the tissue distribution and physiology of xanthine oxidoreductase. Histochem J 1994;26(12):889-95.
- Halliwell B. Free radicals and antioxidants: a personal view. Nutr Rev 1994;1:253-65.
- Kucharsk J, Gvozdjkov A, Herichov I, Gvozdjk J. Significance of nitochondrial Ca2+ transport in ischemic injury and myocardial protection. Bratisl Lek Listy 1994;95(9):391-4.
- Flaherty JT. Reperfusion injury. Free Rad Biol Med 1988;5:409-19.
- Schoenberg MH, Beger HG. Oxygen radicals and postischemic organ damage: pathophysiology, clinical relevance and therapy. Zentralbl Chir 1995;120(3):174-85.
- Ferrari R. Oxygen free radicals at myocardial level: effects of ischemia and reperfusion. Adv Exp Med Biol 1994;366:99-111.
- Hearse D. Xanthine oxidase: a critical mediator of myocardial injury during ischemia and reperfusion? Acta Physiol Scand 1986;(Suppl 548):65-78.
- Buja M, Bick RJ. Control and modulation of neonatal myocyte calcium movements by cytokines. Promega Notes 1994;47:6-9.
- Lapenna D, Mezzetti A, Giola S de, Consoli A, Festi D, Di Llio C, et al. Transmural distribution of antioxidant defences and peroxidation in the rabbit left ventricular myocardium. Pflugers Arch 1994; 427(5-6):432-6.
- Ji LL, Fu RG, Mitchell EW, Griffiths M, Waldrop TG, Swartz HM. Cardiac hypertrophy alters myocardial response to ischemia and reperfusion in vivo. Acta Physiol Scand 1994;151(3):279-90.
- Engelman DT, Watanabe M, Engelman RM, Rousou JA, Kisin E, Kagan VE, et al. Hypoxic preconditioning preserves antioxidant reserve in the working rat heart. Cardiovasc Res 1995;29(1):133-40.
- Aucoin MM, Barhoumi R, Kochever DT, Granger HJ, Burghardt RC. Oxidative injury of coronary venular endothelial cells depletes intracellular glutathione and induces HSP 70 mRNA. Am J Physiol 1995;268(4 Pt 2):H1651-8.
- Luc G, Fruchart J. Oxidation of lipoproteins and atherosclerosis. Am J Clin Nutr 1991;53:206s-9s.
- Feuerstein GZ, Ruffolo RR. Carvedilol, novel multiple action antihypertensive agent with antioxidant activity and the potential for myocardial and vascular protection. Eur Heart J 1995;16(Suppl 7):38-42.
Dr. Félix Broche Valle. Centro de Investigaciones Biomédicas. Instituto Superior de Ciencias Médicas de La Habana. Calle 146 #3102, reparto Cubanacán, municipio Playa. Ciudad de La Habana, Cuba. CP 11600.