










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de Noonan (SN) es una enfermedad monogénica de herencia autosómica dominante con expresión clínica variable.1 Es una de las enfermedades no cromosómicas más frecuentes a nivel mundial, con una incidencia de 1:1000-2500 recién nacidos vivos, por lo que tiene una frecuencia similar a la del síndrome de Down.2 A pesar de esto, se encuentra subdiagnosticada, debido a que presenta gran variabilidad en su presentación clínica. Las características fenotípicas de esta enfermedad se hacen más difíciles de distinguir a medida que aumenta la edad del paciente.3 Sin embargo, un examen físico exhaustivo y la evaluación de los criterios diagnóstico de SN pueden hacer un correcto diagnóstico diferencial de este cuadro con otras alteraciones congénitas, como el síndrome de Turner y los Síndromes neuro-cardio-facio-cutáneos (síndrome de Leopard, Neurofibromatosis Tipo I y síndrome de Williams).3,4
El SN presenta diferentes complicaciones dependiendo del grupo etáreo,5 las más importantes son secundarias a defectos cardiovasculares congénitos, los cuales representan un 50-90 % de complicaciones, y pueden ser leves o graves.6
El objetivo del estudio fue discutir la importancia del examen clínico para su adecuado diagnóstico a partir de las características del síndrome de Noonan en un adulto.
Caso clínico
Paciente de sexo masculino, de 33 años, natural y procedente de la zona rural de Arequipa, Perú. Presenta un cuadro caracterizado por dos meses de fiebre, sudoración nocturna, astenia, hiporexia y disnea a moderados esfuerzos (II/IV NYHA). Fue atendido en un centro de atención primaria donde se le dio tratamiento (no especificado por el paciente ni familiares), con el que el cuadro no remitió y los síntomas se agravaron, se sumaron a los anteriores: edema en miembros inferiores, tos y dolor toráxico. Con estos síntomas es referido a un hospital de nivel III.7
Entre los antecedentes, los familiares refirieron que el paciente nació de parto eutócico, con un peso de 4800 gr, con hallazgos de pie varo derecho, criptorquidia de testículo izquierdo y pterigium coli. No se registró diagnóstico de alguna cromosomopatía o enfermedad genética específica. Durante el período de lactante presentó un desarrollo aparentemente normal, sin problemas para la alimentación e infecciones recurrentes características de SN (otitis media). En el período de la infancia hubo retraso del desarrollo psicomotor, manifestó problemas conductuales, con características de una personalidad hiperactiva, hemorragias con leves traumas y disfonía. No recibió instrucción escolar básica. En la etapa puberal tuvo retraso en el desarrollo de las características sexuales secundarias. En comparación con el resto de niños presentaba dolor torácico, disnea y palpitaciones después de realizar actividades físicas no extenuantes.
En el examen físico se hallaba en aparente mal estado general, regular estado de nutrición, hidratación e higiene. Con una frecuencia cardiaca de 110 latidos por minuto, 20 respiraciones por minuto y presión arterial de 150/60 mmHg. Tenía una talla de 1,50 m y peso de 62 kg. Encontramos piel y conjuntivas pálidas, frente amplia, tejido periorbitario abundante, orejas de implantación baja y rotación posterior, hendidura palpebral antimongoloide, ptosis palpebral, puente nasal deprimido, punta nasal ancha, región malar plana, mejillas prominentes, filtrum profundo (Fig. 1).
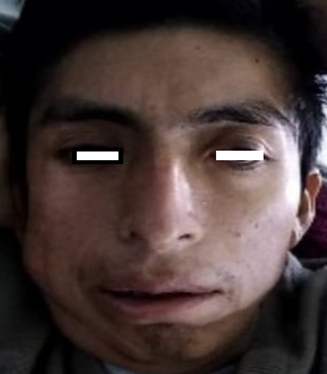
Fig. 1- Rasgos faciales en un paciente con Síndrome de Noonan: Frente amplia, tejido periorbitario abundante, ptosis palpebral hipertelorismo, desviación tipo antimongoloide de fisuras palpebrales, punta nasal ancha, puente nasal deprimido y micrognatia.
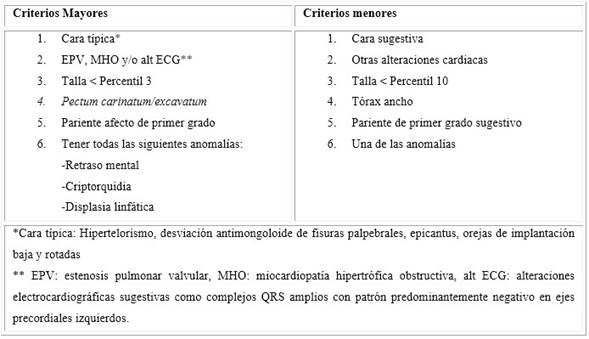
Además de pterigium colli, maloclusión dentaria, dientes pequeños y espaciados, micrognatia, cuello corto, a la inspección de tórax se observó pectus carinatum superior y excavatum inferior (Fig. 2), ingurgitación yugular +++/+++, reflejo hepatoyugular positivo, se auscultó crépitantes bibasales en ambos campos pulmonares, ruidos cardiacos regulares con presencia de soplo diastólico de escape sigmoideo II/VI en foco aórtico. Presentaba, además, una tumoración reductible no dolorosa de 10 a 12 centímetros de consistencia blanda en región inguinoescrotal derecha, criptorquidea de testículo izquierdo y pie varo derecho. Basado en los criterios de diagnóstico clínico de SN el paciente cumple con tres de seis criterios mayores (facie típica, talla adulta inferior a la normalidad 1,60 m en varones, pectus carinatum/excavatum) y dos criterios menores (otras alteraciones cardiacas y pariente de primer grado sugestivo), que concluyen el diagnóstico de dicha enfermedad. En los estudios de laboratorio se encontraron los valores de la Tabla 1.

Fig. 2- Características físicas en tórax de un paciente con Síndrome de Noonan post cirugía de recambio valvular. Pterigium Coli (a), pectus carinatum superior (b) y excavatum inferior (c). Cicatriz post quirúrgica de exéresis y recambio valvular.
El electrocardiograma mostró una frecuencia de 105 latidos por minuto, intervalos PR y QT conservados, eje eléctrico del corazón en 60˚, y signos característicos de hemibloqueo anterior izquierdo y taquicardia sinusal.
En la radiografía observamos aumento de la trama vascular y no se hallaron alteraciones del parénquima pulmonar. En el hemocultivo se aisló Staphylococcus aureus, El estudio ecocardiográfico reveló cardiomegalia, función sistólica y diastólica de ventrículo izquierdo disminuida, con fracción de eyección de 46 %, vegetación en válvula aórtica compatible con endocarditis infecciosa, asociada a insuficiencia aórtica severa y derrame pericárdico leve a moderado.
Ante estos hallazgos, la conclusión diagnóstica fue endocarditis infecciosa subaguda con insuficiencia cardiaca congestiva, anemia moderada y hernia inguinal derecha reductible. El tratamiento inicial fue: vancomicina 1g/12 h, gentamicina 160 mg/24h, furosemida 20 mg/12h, enalapril 10 mg/12h, espironolactona 25 mg/24h, bisoprolol 1.25 mg/24h. Sin embargo, el paciente abandonó el hospital al quinto día de iniciado el tratamiento dejándolo inconcluso.
Dos meses después, el paciente reingresó por el servicio de emergencia, descompensado hemodinámicamente, con disnea en reposo (IV/IV Escala NYHA), edemas en miembros inferiores (+++/+++) y oliguria, por lo cual se decidió su internamiento. Los exámenes auxiliares (Tabla1) revelaron anemia, plaquetopenia y microalbuminuria. El informe ecográfico mostró esplenomegalia, derrame pericárdico y pleural bilateral.
Tabla 1- Resultados de los exámenes de laboratorio, se comparan los valores del primer y segundo ingreso

Considerando el mal pronóstico y el grado de resolución del nosocomio, se decidió tratar la endocarditis para luego referirlo a un centro hospitalario de mayor complejidad, en el cual se le realizó una exéresis y recambio valvular. La cirugía fue realizada de forma exitosa en un centro especializado de la ciudad de Lima; sin embargo, el paciente no fue llevado a controles posteriores y falleció debido a sobreanticoagulación.
Discusión
El SN se encuentra incluido en el listado de “Enfermedades Raras o Huérfanas” según el Ministerio de Salud (MINSA).8 Según el Instituto Nacional de Salud del Niño, representa el 0,91 % de trastornos genéticos diagnosticados en esta entidad.9) Es una enfermedad desatendida en nuestro medio, a pesar de existir protocolos internacionales que proporcionan información del diagnóstico y seguimiento. Esta enfermedad se produce por mutaciones heterocigotas en genes de la vía RAS/MAPK, principalmente el gen PTPN11, el cual se localiza en el brazo largo del cromosoma 12 (12q24.1) y codifica la tirosina fosfatasa no receptora (SHP2), que intervienen en la traducción de diversos factores de crecimiento, hormonas y vías de señalización de citocinas de control, las cuales se encuentran implicadas en un amplio rango de enfermedades denominadas síndromes neuro-cardio-facio-cutáneos. Estos síndromes comparten características clínicas como: talla baja, cardiopatía y dismorfia facial.10,11,12 A pesar de compartir dichas características clínicas, no es necesario pruebas moleculares para establecer el diagnóstico del SN, el cual se fundamenta en los criterios clínicos expuestos en la tabla 2. El diagnóstico clínico definitivo se realiza si el paciente cumple: 2 criterios mayores, 1 mayor y 2 menores o 3 menores.2,5 En el presente caso se encontraron tres criterios mayores y 2 menores (1, 3 y 4 - 5 y 6 respectivamente de la Tabla 2).
Existen características clínicas variables que se encuentran ausentes en el SN, las cuales son de utilidad para realizar el diagnóstico diferencial del resto de síndromes neuro-cardio-facio-cutáneo. Las pruebas moleculares solo son necesarias en casos límites o dudosos, y un resultado negativo no descarta el diagnóstico del SN.12
Las guías y protocolos de atención y manejo de SN sugieren que, de existir sospecha clínica de este síndrome, debe tratarse de hacer el diagnóstico tempranamente, con el propósito de realizar un seguimiento multidisciplinario que evite complicaciones, las cuales varían según el grupo etario del paciente.4) En nuestro caso el diagnóstico fue tardío y cuando las complicaciones estructurales dieron origen a sobreinfección, que resultó en una mala evolución del cuadro.
Entre las complicaciones cardiovasculares se ha reportado estenosis pulmonar valvular (50 %), defectos septales auriculares (10 %), defectos septales ventriculares (5 %), ductus arterioso persistente (3 %) y con menos frecuencia estenosis aórtica y anomalías mitrales.6 Creemos que, a pesar de ser poco frecuente, la estenosis aórtica pudo ser la alteración estructural que favoreció el desarrollo de la endocarditis en el paciente. A pesar de que el paciente presenta una historia compatible con esta sospecha, no tenemos estudios ecocardiográficos previos que confirmen la presencia de esta estenosis.
A pesar de que se estima que más de dos millones de peruanos tienen una enfermedad genética,13 en el Perú no existe aún un sistema eficiente de registro de la información sanitaria, ni políticas de inversión científico tecnológicas para el diagnóstico específico de las condiciones genéticas o el seguimiento adecuado de estos pacientes. En nuestro caso la falta de adherencia al tratamiento y la dificultad para el seguimiento y control del paciente fueron determinantes en el desenlace del mismo. Esto evidencia la importancia de los factores sociales y el manejo multidisciplinar que estos casos requieren.
Como limitación del presente reporte podemos mencionar que no se pudieron realizar pruebas moleculares para identificar las mutaciones de los genes MAPK21, PTPN11, KRAS, SOS1, 6RAF1, BRASF, NRAS y RIT1, lo que hubiera reforzado el diagnóstico de la enfermedad. La falta de un adecuado control y documentación de la evolución del paciente fue otro limitante para poder hacer un seguimiento a su historia.
Conclusiones
El SN puede ser diagnosticado en base al examen clínico y los criterios diagnósticos. El diagnóstico temprano y el estudio de las complicaciones cardiovasculares son necesarias para un adecuado tratamiento.
Se cuenta con la autorización del paciente para la presentación del caso, además las autoridades del hospital otorgaron los permisos para su publicación.

