










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Ortopedia y Traumatología
versión On-line ISSN 1561-3100
Rev Cubana Ortop Traumatol v.2004 n.2 Ciudad de la Habana jul.-dic. 2004
Hospital Ortopédico Docente "Fructuoso Rodríguez" Hospital Pediátrico Docente "Ángel Arturo Aballí"
Ciudad de La Habana, Cuba
Fractura patológica en edad pediátrica a causa de displasia fibrosa ósea
Dr. Lázaro Robaina Ruiz1 y Dr. Arturo Pablo Vega Ojeda2
Robaina Ruiz L. y Vega Ojeda AP. Displasia fibrosa ósea como causa de fractura patológica en edad pediátrica. Rev Cubana Ortop Traumatol 2004;18(2).
Resumen
Se presenta un paciente de 13 años de edad, que a causa de un traumatismo sobre la cadera derecha sufrió fractura del extremo proximal del fémur. Se hace una revisión bibliográfica sobre clasificación actual, signos clínicos, radiográficos y anatomopatológicos sobre displasia fibrosa ósea, Se halló en los exámenes clínicos y paraclínicos del paciente que la zona fracturada comprendía hueso patológico. Se muestran las radiografías con los hallazgos obtenidos. Se realizó bopsia , curetaje de la lesión y se rellenó con material biocompatible. Constituye el primer caso reportado en Cuba de fractura en hueso patológico del extremo proximal del fémur, por displasia fibrosa ósea en edad pediátrica.
Palabras clave: displasia fibrosa ósea, extremo proximal del fémur, curetaje, implante biocompatible, fractura en hueso patológico .
La displasia fibrosa ósea está considerada conceptualmente como una lesión pseudotumoral benigna del esqueleto en crecimiento, en la que los elementos óseos son normales pero no muestran signos de diferenciación hacia estructuras maduras.1 Tiene como sinonimia, tumor de Lichtenstein y Jaffe, y según algunos autores es más frecuente en la infancia y en la adolescencia. Numerosas anomalías pueden asociársele como: pigmentación anormal de la piel, desarrollo sexual precoz, maduración esquelética temprana, mixoma intramuscular, hipertiroidismo, enfermedad de Cushing y osteomalacia hipofosfatémica.1,2
Posee 3 formas clínicas de presentación: monostótica ( afecta un solo hueso), poliostótica ( afecta varios huesos pero nunca a todos), poliostótica tipo síndrome de Albright (asociada con manchas pigmentadas "café con leche" y alteraciones endocrinas, sobre todo, pubertad precoz).1-4
La forma monostótica comprende el 70 % de los casos, es igual de frecuente en ambos sexos, generalmente aparece en los primeros años de la adolescencia, suele dejar de crecer cuando cierran las epífisis. Las localizaciones más frecuentes, en orden descendente, son: costillas, fémur, tibia, mandíbula, bóveda craneal y húmero. Con frecuencia es asintomática, y muchas veces se descubre accidentalmente. Puede producir gran deformidad, y si afecta el esqueleto craneofacial es posible la desfiguración del paciente. No evoluciona hacia la forma poliostótica.
La forma poliostótica sin disfunción endocrina ( 27 %) aparece en sujetos más jóvenes que en la forma monostótica y puede causar problemas hasta bien entrada la vida adulta. Las localizaciones más frecuentes, en orden descendente, son : fémur, cráneo, tibia, húmero, costillas, fíbula, radio, ulna, mandíbula y cinturas pélvicas y escapular. Es posible la afectación craneofacial y la pelvicoescapular causa graves deformidades invalidantes como la deformidad en cayado de pastor del fémur proximal.
La forma poliostótica con disfunción endocrina (Síndrome de Mc Cune-Albright), comprende el 3 % de los casos. Las endocrinopatías se deben a una mutación somática "no hereditaria" que se produce durante la embriogénesis, y pueden corresponder con pubertad precoz, hipertiroidismo, adenomas hipofisarios secretores de hormona del crecimiento e hiperplasia suprarrenal primaria con Síndrome de Cushing. La mutación afecta el gen que codifica una proteína estimuladora de unión al nucleótido de guanina y conlleva un exceso de producción de adenosina monofosfato cíclico, con la consiguiente hiperfunción glandular endocrina. La presentación clínica más frecuente es el desarrollo sexual precoz y afecta más a las mujeres. Las lesiones óseas son a menudo monolaterales y la pigmentación cutánea suele limitarse al mismo lado del cuerpo. Las máculas cutáneas son grandes, de color oscuro "café con leche" con bordes serpiginosos, como la costa de Maine en EE.UU. Se localizan en cuello, tórax, espalda, hombro y región pélvica.1,2,4
Mucho se ha especulado acerca de la posibilidad de transformación maligna de la enfermedad y aunque es un hecho raro, es una realidad ya sea con radioterapia previa o sin ella. Para la forma monostótica la incidencia es de 0,4 % y para la forma poliostótica, 4%.5 Casi siempre la transformación maligna ocurre en la edad adulta, sobre todo, después de la radioterapia, por lo cual está formalmente contraindicada. Los tumores malignos más frecuentemente desarrollados son osteosarcomas, fibrosarcomas, condrosarcomas, histiocitoma fibroso maligno y se ha reportado degeneración quística.1-3,5
Anatomía patológica
Las lesiones son de localización intramedular, bien circunscritas y de tamaño variable, las más grandes se expanden y deforman el hueso El tejido es de color blanco tostado, granujiento, formado por trabéculas curvas, de hueso "no laminar", rodeadas por proliferación fibroblástica, comparables con letras o caracteres chinos. El hueso carece de ribetes osteoblásticos; hay nódulos de cartílago hialino, con aspecto de placa de crecimiento desorganizada. La degeneración quística, la hemorragia y los macrófagos espumosos son hallazgos frecuentes, también se han descrito depósitos de colesterol. En general, se presentan grandes áreas quísticas e incluso regiones de formación cartilaginosa. 1,2,.6
Datos radiográficos
La apariencia radiográfica típica muestra osteólisis extensa, en forma de burbujas de jabón, con bordes escleróticos bien delineados. Hay expansión del hueso y afinamiento de la cortical con numerosas áreas quísticas trabeculadas.5 Según algunos autores, el aspecto radiográfico puede sugerir en ocasiones un problema más ominoso y se hace necesaria la biopsia para establecer el diagnóstico.2 La forma monostótica es más sencilla de diagnosticar mediante rayos x por su aspecto de vidrio esmerilado y sus márgenes bien definidos.1
Presentación del caso
Paciente masculino, de piel blanca y 13 años de edad, que nació de parto eutósico, a término, con buena vitalidad, sin trauma obstétrico ni alteraciones durante la gestación. La historia familiar no recoge datos de otros tumores ni fracturas en hueso patológico. Desarrollo psicomotor normal.
En una competencia deportiva, el paciente sufrió una caída sobre la cadera derecha y recibió intenso trauma sobre ella, luego de lo cual no pudo continuar la actividad a causa de un gran dolor e imposibilidad para caminar. Fue trasladado al Cuerpo de Guardia donde se le realizó examen físico que arrojó impotencia funcional absoluta del miembro inferior derecho, dolor espontáneo y provocado a los movimientos pasivos y activos, aumento de volumen de la raíz del muslo, palpación dolorosa superficial y profunda sobre la zona del trocánter mayor de la cadera derecha. Se le practicaron radiografías de la pelvis ósea en su conjunto y de ambas caderas, que constataron fractura del extremo proximal del fémur derecho en la región intertrocantérica en un área que impresionaba como hueso patológico (figs. 1 A, y B).

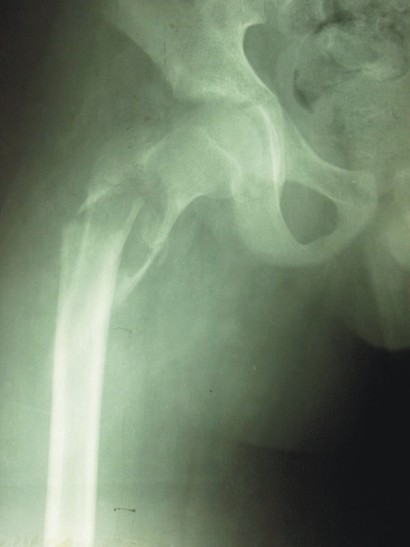
FIGS. 1 A y B). Radiografías anteroposteriores de pelvis ósea donde se observa fractura del extremo proximal del fémur derecho, ubicada en el macizo intertrocantérico, con desplazamiento escaso. El hueso posee evidentes características patológicas.
Se le realizó examen hemoquímico general que comprendía: hemograma con diferencial, uroanálisis, fosfatasa alcalina y ácida, coagulación y sangramiento, velocidad de sedimentación globular y glicemia y creatinina, todos con resultados de valores normales.
Con los citados hallazgos clínicos y radiográficos se decidió llevar el paciente al quirófano y realizar la biopsia previa correspondiente. Se recibió el resultado y se concluyó displasia fibrosa ósea del extremo proximal del fémur derecho, por lo que se decidió realizar la cirugía definitiva, que consistió en curetaje del hueso patológico y relleno con material biocompatible del tipo de la hidroxiapatita porosa (microporos de 200 micras) (figs. 2 A y B). Se le colocó hemiespica toracopédica posoperatoria, por espacio de 8 semanas, y se comenzó luego la rehabilitación, a la semana 16 comenzó el apoyo. La marcha la realizó sin dolor y no fue claudicante, sin signo de Trendelemburg. Se siguió por consulta por espacio de 2 años y se evidenció una evolución excelente (figs. 3 A y B).


FIGS. 2 A y B). Radiografías anteroposteriores de la pelvis ósea y del macizo trocanteriano afectado. Se observa el material biocompatible implantado e incorporado, un adecuado afrontamiento de las superficies de la zona fracturada y el mantenimiento de la integridad de la pared posteromedial del cálcar femoral con el fin de lograr estabilidad de la zona para el apoyo posterior.

FIGS. 3. A y B). Zona de abordaje lateral estricto para el extremo proximal de la
diáfisis del fémur. Paciente en bipedestación normal posterior al tratamiento.
Discusión
La tendencia actual de tratamiento en la displasia fibrosa ósea constituye un aspecto controversial, ya que no todas las escuelas poseen iguales enfoques. Para unas, el tratamiento quirúrgico está indicado cuando exista deformidad significativa, fractura en hueso patológico y dolor considerable. La escuela de Campbell recomienda de elección, el curetaje e injerto óseo. Para las deformidades se recomiendan las osteotomías con fijación interna; para las fracturas, el curetaje de la lesión con injerto óseo y fijación interna de la fractura, aunque es cierto también que la mayoría de los pacientes con esta afección no requieren tratamiento quirúrgico.
En pacientes pediátricos con la enfermedad biológicamente activa es poco prudente intentar un injerto de hueso, debido a la rápida desaparición del material injertado y también a la actividad continua del proceso patológico. Por ello, para algunos autores, los objetivos del tratamiento en pacientes pediátricos debe ser la prevención y el tratamiento de una deformidad, en especial en la extremidad inferior.2 Incluso en la edad adulta, la mayoría de los hombres y mujeres con displasia fibrosa no requieren cirugía. Por otra parte, en adultos, el mejor tratamiento quirúrgico consiste en practicar el injerto de hueso largo autógeno con cuñas de fíbula, combinada con injerto de hueso esponjoso autógeno. Este método muestra una tasa de éxito más elevada en el grupo de adultos que en el grupo pediátrico.7 Otro dato importante es que la fijación metálica per se, conduce a una incidencia elevada de fracasos debido a la escasez de materia prima ósea.2 También se plantea que cuando se realiza la fijación interna debe ser colocada por tiempo indefinido.4 Todos los reportes condenan unánimemente el empleo de la radioterapia como método de tratamiento, porque puede producir degeneración sarcomatosa inducida por radiaciones.1-6
Concluyendo se puede afirmar que la displasia fibrosa ósea es una afección que posee varias formas de presentación, de lo que dependerá el pronóstico y la evolución de los pacientes. En la forma monostótica es mucho más fácil de diagnosticar que en las poliostóticas y nunca evoluciona hacia ellas. Las formas poliostóticas pueden estar o no acompañadas de endocrinopatías y la más frecuente es la pubertad o desarrollo sexual precoz..
Todos los pacientes con la alteración de referencia deben ser seguidos en consulta hasta que arriben a la vida adulta.. En los pacientes muy pequeños, la gran actividad biológica del proceso puede constituir una contraindicación a la cirugía, excepto en aquellos casos en que se produzcan fracturas en hueso patológico o deformidades groseras de gran importancia.
El paciente que se presenta es el primer caso reportado en Cuba por fractura en hueso patológico del extremo proximal del fémur por displasia fibrosa ósea, en edad pediátrica.
Summary
A 13-years old male patient who suffered fracture of the proximal end of femur due to right hip trauma is presented in this paper. A literature review was made on current classification, clinical, radiographic and anatomopathological signs of bone fibrous dysplasia. In the clinical and paraclinical tests practiced to the patient, it was found that the fractured area included a pathological bone. Radiographies with the findings observed are shown. Biopsy, curettage of lesion and filling with biocompatible material were performed. This is the first case of fracture in pathological bone of the proximal end of femur from osseous fibrous dysplasia at pediatric age in Cuba
Key words: osseous fibrous dysplasia, proximal end of femur, curettage, biocompatible implant, fracture in pathological bone.
Résumé
Un patient âgé de 13 ans atteint d'une fracture de la partie proximale du fémur à cause d'un traumatisme de la hanche droite, est présenté. Une revue bibliographique sur la classification actuelle, les signes cliniques, radiographiques et anatomopathologiques de la dysplasie fibreuse des os est effectuée. Dans les examens cliniques et paracliniques du patient, on a trouvé que l'aire de la fracture comprenait l'os pathologique. Les clichés montrent les résultats obtenus. Une biopsie et un curetage de la lésion ont été réalisés, et elle a été remplie d'un matériel biocompatible. C'est le premier cas rapporté à Cuba de fracture de l'os pathologique de la partie proximale du fémur par dysphasie fibreuse des os en âge pédiatrique.
Mots clés: dysplasie fibreuse des os, partie proximale du fémur, curetage, implant biocompatible, fracture de l'os pathologique.
Referencias bibliográficas
- Cotran SR, Kumar V, Robbins LS. Patología estructural y funcional. 5a ed. Madrid : Mc Graw-Hill Interamericana; 1996:1362-4.
- Campbell WC. Cirugía Ortopédica..V1. 8a ed. Buenos Aires: Editorial Médica Panamericana; 1994 p. 234-4.
- Herring JA. Tachdjian´s Pediatric Orthopaedics. V3. 3a ed. Philadelphia: Saunders; 2002 p. 1909-12.
- Staheli LT. Fundamentals of Pediatric Orthopaedics. 2a ed. Philadelphia: Lippincott-Raven; 1998 p. 137-9.
- Yabut S, Kenar S, Sissons H . Malignant transformation of fibrous dysplasia. Clin Orth Rel Res. 228: 81.
- Mercer W, Duthie RB. Orthopaedic surgery. 6a ed. London: Arnold; 1964 p. 144-5.
- Enneking WF, Gearent PF. Fibrous Dysplasia of the femoral neck. Treatment with cortical bone grafting. JBJS 68 A: 1415.
Recibido: 10 de mayo de 2004. Aprobado: 12 de abril de 2004.
Dr. Lázaro Robaina Ruiz. Hospital Pediátrico Docente "Angel Arturo Aballí". Calzada de Bejucal km 7 ½. Arroyo Naranjo, Ciudad de La Habana, Cuba.
1 Especialista de I Grado en Ortopedia y Traumatología. Instructor de la Facultad "Calixto García".
2 Especialista de I Grado en Ortopedia y Traumatología. Instructor de la Facultad "Calixto García".

