










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Archivo Médico de Camagüey
versión On-line ISSN 1025-0255
AMC vol.8 no.3 Camagüey mayo.-jun. 2004
ARTÍCULOS ORIGINALES
Caracterización molecular de dos pacientes 46 XY, fenotipo femenino. Síndrome de Swyer
Molecular characterization of two patients 46XY: feminine phenotype. Swyer syndrome
Dr. Juan Carlos Piña Napal; Dr. Charles T. Vázquez Drake; Dra. Hilda Granda Ibarra; Lic. Blanca Suardíaz Martínez
Hospital Pediátrico Provincial Docente de Camagüey. Departamento Provincial de Genética Médica.Camaguey, Cuba.
RESUMEN
El síndrome de Swyer es un desorden de la diferenciación sexual en el cual los pacientes exhiben un fenotipo femenino con cariotipo 46 XY e hipoplasia gonadal sin células germinales. Dentro de las posibles causas para esta reversión sexual se encuentra la ausencia del gen SRY ubicado en el cromosoma Y. Éste ejerce el papel rector en la cascada de genes que intervienen en la diferenciación sexual. En este trabajo se utilizó la técnica de reacción en cadena de la polimerasa para amplificar el gen SRY, con el fin de conocer si la ausencia del mismo podía explicar la reversión sexual en nuestros dos pacientes. Se detectó la presencia del gen SRY y concluimos que la causa del síndrome puede estar dada por mutaciones en el gen SRY o en otros que participan en la cascada de diferenciación sexual en humanos.
DeCS: DISGENESIA GONADAL 46 XY; ANOMALÍAS DE LOS CROMOSOMAS SEXUALES.
ABSTRACT
Swyer syndrome is a disorder in sexual differentiation in which patients show a femenine phenotype with 46xy karyotype and gonadal hypoplasia without germinal cells. Among the possible causes for sexual reversion present in this syndrome it is the absence of SRY gene, located in the chromosome Y. This exerts the rector role in the gene cascade which intervenes in the sexual differentiation. In this work we performed the technique of chain reaction of polymerase for enlarging the SRY gene, with the aim of knowing if the abscense of it may explain the sexual reversion in our two patients. We detected this presence of SRY gene and concluded that the cause of this syndrome may be mutations in the SRY gene or in other genes that participate in the sexual differentiation cascade in humans.
DeCS: 46 XY GONADAL DYSGENESIS; SEX CHROMOSOME ABNORMALITIES.
INTRODUCCIÓN
La disgenesia gonadal pura 46 XY, también conocida como síndrome de Swyer, es un desorden de la diferenciación sexual en el cual los pacientes exhiben un fenotipo femenino con cariotipo 46 XY e hipoplasia gonadal sin células germinales. 1 Al nacimiento parecen niñas normales e incluso puede haber presencia bilateral de genitales internos femeninos, 2 sin embargo, no mestrúan al llegar a la pubertad con involución prematura de los ovarios resultantes en vestigios gonadales no funcionales y compuestos por tejido fibroso con una alta incidencia de neoplasias malignas como es el caso del germinoma. En la infancia especialmente, el más frecuente es el gonadoblastoma (carcinoma in- situ) con un riesgo calculado de un 30 % aproximadamente. 3, 4 El síndrome se caracteriza además por tener cromatina negativa, estatura normal sin estigma de Síndrome de Turner, excepto por supuesto la carencia de caracteres sexuales secundarios y los vestigios gonadales. 5
Como la determinación del sexo tiene lugar durante los procesos de morfogénesis y diferenciación celular, la búsqueda y el estudio de la causa genética del síndrome que nos ocupa se hacen un poco más complejos, pues como se sabe, estos procesos a su vez son dependientes de la actividad de un grupo de genes que interactúan de forma entrecruzada en una vía metabólica, y además las vías que permiten el desarrollo de un varón o una hembra están influenciadas por diferentes condiciones y mecanismos moleculares que determinan cuál de las vías será seleccionada. 6, 7
En los mamíferos hay un gen ubicado en el cromosoma Y que es el encargado de establecer el control en la diferenciación sexual. 8 Este gen tiene una acción dominante en la determinación del varón. 9 El evento central en esta especie es la diferenciación en testículos en lugar de ovarios a partir de las gónadas indiferenciadas, 10 el resto de las diferencias entre los sexos son secundarias a los efectos hormonales o a los factores producidos por las gónadas, razón por la cual se explica que la determinación del sexo es equivalente a la determinación del testículo.
En los humanos la región determinante del sexo en el cromosoma Y, ha sido delimitada a una secuencia de 35 kilobases (kb), cercana a la frontera de la región pseudoautosómica. En esta región se encuentra ubicado el gen SRY (Sex-determining region of the Y chromosome), ubicado en Yp1, región 1A1. Es específico de este cromosoma, se conserva en todos los mamíferos 8, 10 y tiene un papel rector en la cascada de genes que intervienen en la diferenciación sexual. 11 Es conocido que este gen de 4 737 pares de bases, codifica para una proteína nuclear de 204 aminoácidos. 12 Ésta pertenece a la familia de las proteínas interruptores o Switch proteínas, 13 cuya función en el ratón ha sido asociada con la unión en el ADN a elementos de control de la expresión genética (DNA biding Protein). 8 En humanos el producto de la expresión del SRY es un factor transcripcional 14 que tiene como blanco los promotores de una cascada de genes que determinan la diferenciación de las gónadas masculinas, 15 cuya función es modulada por una PKA (cyclic AMP-dependent protein kinase), es decir, por una proteína quinasa dependiente de AMP-cíclico.
El objetivo de esta investigación es determinar la ausencia del gen SRY para explicar el comportamiento del síndrome de Swyer en nuestros pacientes, sobre todo al considerar que éste dirige la determinación del sexo en los mamíferos.
MÉTODO
Se empleó sangre total de dos pacientes voluntarios con síndrome Swyer, provenientes de la consulta general de endocrinología del Instituto Nacional de Endocrinología y Enfermedades Metabólicas de Ciudad de La Habana. Para la extracción de ADN utilizamos el método de precipitación salina de Salting Out, reportado por Miller y cols., modificado. 16 El ADN fue chequeado por electroforesis aplicando 1µl de la solución de ADN en gel de agarosa normal 1 % con 0,5 µg/ml de bromuro de ethidium para asegurar la tinción del ADN; se visualiza con la ayuda de un transiluminador ultravioleta. Este procedimiento, al igual que el espectrofotométrico, se empleó en todas las ocasiones que fue necesario visualizar el ADN y chequear su concentración.
Se preparó la mezcla de reacción para realizar la reacción en cadena de la polimerasa (PCR, Polymerase Chain Reaction) donde las condiciones fijadas fueron desnaturalización a 94º C por 10 min inicialmente y luego 30 ciclos del siguiente modo:
-. 94º C por 45 seg.
-. 68º C por 2 1/2 min.
Se depositó en un tubo eppendorf 12,5 µl del producto del PCR, se añadieron 2 µl de Buffer NeB-2 [10X]; 0,4 µl de albúmina de suero bovino (BSA) [10 mg/ml]; 4,1 µl de H2O destilada estéril y finalmente, 1 µl de la enzima de restricción Xmn I [6U/µl]; para un volumen total de reacción de 20 µl. Después se colocó en baño de María a 37° C durante 3 h para asegurar la digestión completa.
El producto del PCR digerido se corrió en un gel de poliacrilamida al 10 %, desnaturalizante, para ello se utilizó una cámara de electroforesis adecuada. Se corrió durante 3 h a voltaje constante de 250 V y corriente de 54 mA. En este proceder, se aplicó el Marker V (Plásmido pBR322 cortado con la enzima de restricción Hae III) como patrón de peso molecular.
Para visualizar las bandas en el gel de poliacrilamida se procedió al método de tinción con plata recomendado por la firma Perkin Elmer. (Se sumergió el gel de poliacrilamida en metanol al 40 % en agitación por 10 min, después se sumergió por 6 min. en ácido nítrico [160 mM]. Una vez tratado el gel con ácido nítrico se sumergió en agua bidestilada por 5 min. para después proceder a sumergir en nitrato de plata [12 mM], se volvió a enjuagar en agua bidestilada por 5 min, y por último, se sumergió el gel en solución de desarrollo a 4 °C (Carbonato de Sodio [280 mM], formaldehído 37 %) hasta que comenzaron a aparecer teñidas las bandas, la reacción se detuvo con solución de ácido cítrico [1M], para que no aumentara mucho el fondo).
RESULTADOS
A partir de las muestras de sangre que se extrajeron de cada paciente se logró purificar cantidad suficiente de ADN genómico. Fueron chequeadas en el espectrofotómetro, su concentración promedio fue de 11.9 µg/ml con absorvancia de 0.239 AU, 93 % de pureza y 0 % de proteínas, para el paciente número 1; y 9,2 µg/ml con absorvancia de 0.183 AU, 92 % de pureza y 0 % de proteínas, para el paciente número 2. Se chequearon además por electroforesis en gel de agarosa normal al 1 %, teñidas con bromuro de etidium y visualizadas en un transiluminador ultravioleta, con lo cual se verificó que el ADN no estaba degradado.
Se realizó la técnica de PCR en 30 ciclos. Su producto fue digerido con la enzima de restricción Xmn I y al examinar el gel de poliacrilamida se observó en los dos pacientes (pozos seis y siete del gel de poliacrilamida) la presencia de cuatro bandas. Una con talla de 510 Pb, correspondiente al fragmento del gen Apo B-100 100 (Apolipoproteína B-100), como control interno de amplificación, y tres adicionales: una de 399 pb, otra de 107 pb y otra 103 de pb., correspondientes a la digestión del fragmento del gen SRY de 609 Pb (Figura 1).
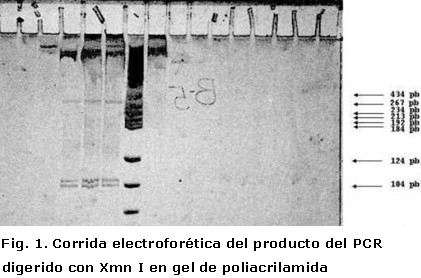
En el cuarto pozo del gel de poliacrilamida fue aplicado como control el producto del PCR de un varón normal, se mostraron sus dos bandas: una superior de 609 pb que se corresponde con el fragmento amplificado del gen SRY y una inferior de 510 pb que se corresponde con el fragmento amplificado del gen Apo-B100 (el gen Apo-B 100 se encuentra en 2p23-2p24 y por tanto, está presente en hembras y en varones). En el pozo nueve fue aplicado como control el producto del PCR de una hembra normal que mostró una única banda de 510 pb que se corresponde con el fragmento amplificado del gen Apo-B100, pues en ésta no debe aparecer el fragmento del gen SRY que una hembra normal no posee.
En los pozos cinco, seis y siete fueron aplicadas las muestras digeridas con la enzima Xmn I de un varón normal, el paciente número 1 y el paciente número 2, respectivamente. Se observaron cuatro bandas que corresponde, desde arriba hacia abajo, con el fragmento amplificado del gen Apo-B100 de 510 pb y tres bandas más, de 399,107 y 103 pb que corresponden a los fragmentos generados por la digestión de la enzima Xmn I sobre el fragmento amplificado del gen SRY, este último fue digerido con la enzima Xmn I, como confirmación de que efectivamente estábamos en presencia de la secuencia del gen SRY que deseábamos amplificar. (Xmn I tiene dos sitios de restricción en la secuencia de amplificada del gen SRY y no reconoce sitio alguno sobre la secuencia amplificada del gen Apo-B100). Finalmente en el pozo número ocho aplicamos el Marker V como patrón de peso molecular.
DISCUSIÓN
Al examinar los resultados lo primero que advertimos fue la presencia del gen SRY en nuestros pacientes, lo cual constituye un hallazgo de importancia si tenemos en cuenta que la ausencia de este gen podría explicar por sí sola la causa de la reversión sexual en nuestros pacientes, pues se conoce que puede haber perdidas de genes a partir del intercambio de material genético entre el cromosoma (Y) y el cromosoma (X). 1 Recientemente se ha aislado y caracterizado un gen en el cromosoma (Y), el PRKY que se sabe tiene una homología muy grande con un gen previamente aislado del cromosoma (X) en Xp22.3, el PRKX, este representa un miembro de la familia de las Proteínas Quinasas dependientes de AMP-cíclico para los residuos de serina y treonina. En el 35 % de los pacientes XX con fenotipo masculinos y XY con fenotipo femeninos se ha demostrado que el intercambios entre Xp/Yp ocurre en cualquier parte próximo al SRY y tiene lugar entre PRKY y PRKX. La gran homología y la misma orientación explican la alta frecuencia de apareamientos anormales y las subsecuentes recombinaciones ectópicas. Es precisamente en el intercambio de este material genético entre el cromosoma (X) y el cromosoma (Y) que puede ocurrir la perdida del gen SRY. 17
Como no es la ausencia del gen SRY la causa de la reversión sexual en nuestros pacientes, pensamos que una posible causa podrían ser las mutaciones en el gen SRY propiamente dicho, como ha sido reportado en la literatura internacional. En la misma se asegura que entre un 10 y 15 % de los pacientes con el síndrome de Swyer tienen mutaciones en el gen SRY, donde las deleciones e inserciones representan el 20 %. 1, 12, 14 Es justo referir también que se conocen muchos casos de reversión sexual que no son explicados por alteración en el gen SRY, 18 pues se han reportado mutaciones en otros genes que están involucrados en la cascada de diferenciación sexual, ubicados en el cromosoma (X) o en cromosomas autosómicos como el 9p24.1, 10q25 19 quizás con la excepción del gen SOX9, al menos sin malformaciones del esqueleto como ocurre en nuestros pacientes. 20 es probable también que éste es el caso de los pacientes que estudiamos aquí, pero en muestro medio no podemos demostrarlo.
CONCLUSIONES
Se determinó por P.C.R. y restricción que nuestros pacientes 46 XY fenotipo femenino son portadores del gen SRY. El origen de las anormalidades fenotípicas en nuestros pacientes están dadas por mutaciones en el gen SRY o en otros genes que intervienen en la cascada de diferenciación sexual.
REFERENCIAS BIBLIOGRÁFICAS
1. Scriver CR. The metabolic and molecular bases of inherited disease. Vol. 1. 7 ed. New York; 1995.
2. Nagai K. Molecular evolution of Sry and Sox gene. Gene. 2001;270(1-2):161-9.
3. Salas Cortes L, Jaubert F, Bono MR, Fellous Ml. Expression of the human SRY protein during development in normal male gonadal and sex-reversed tissues. J Exp Zool. 2001;290(6):607-15.
4. Koopman P, Bullejos M, Bowles J. Regulation of male sexual development by Sry and Sox9. J Exp Zool. 2001; 290(5): 463-74.
5. Veitía RA, Salas-Cortes L, Ottolenghi C, Pailhoux E. Testis determination in mammals: more questions than answers. Mol Cell Endocrinol. 2001;179(1-2):3-16.
6. Larsen WJ. Human embryology: development of the urogenital system. 2 ed. St. Louis:Mosby;1997.
7. Ikeda Y. The genes in the molecular cascade of the mammalian sexual development. Nippon-Rinsho. 1997;55(11):2809-15.
8. MaClean HE, Warne GL, Zajac D. Intersex disorders: shedding light on male sexual differentiation beyond SRY. Clin-Endocrinol-Oxf. 1997;46(1):101-8.
9. Baxevains AD, Landsman D. The HMG-1 box protein family: classification and functional relationships. Nucleic Acids Research. 1995;23(9):1604-13.
10. Werner MH, Juth JR, Gronenborn AM, Clore GM. Molecular basis of human 46 XY sex reversal revealed from the three-dimensional solution structure of the human SRY-DNA complex. Cell. 1995;81(5):705-14.
11. Lovell Badge R, Hacker A. The molecular genetics of SRY and its role in mammalian sex determination. Philos-Trans-R-Soc-Lond-B-Biol-Sci. 1995; 350(1333):205-14.
12. Coutin AS, Hamy A, Fondevilla M, Savigny B, et al. Pure 46XY gonadal dysgenesis. J-Gynecol- Obstet-Biol-Reprod-Paris. 1996; 25(8): pp 792-6.
13. Hines RS, Tho SP, Zhang YY, Plouffe LJr. Paternal somatic and germ-line mosaicism for a sex-determining region on Y (SRY) missense mutation leading to recurrent 46,XY sex reversal. Fertil Steril. 1997;67(4):675-9.
14. Domenice S, Yumie Nishi M, Correia Billerbeck A. A novel missense mutation (S18N) in the 5' non-HMG box region of the SRY gene in a patient with partial gonadal dysgenesis and his normal male relatives. Hum Genet. 1998;102(2):213-5.
15. Ion R, Telvi L, Chaussain JL, Barbet JP. Failure of testicular development associated with a rearrangement of 9p24.1 proximal to the SNF2 gene. Hum Genet. 1998;102(2):151-6.
16. Miller S . Método de extracción de ADN por salting out. Nucleic Acids Research. 1998;16(3):1215.
17. McElreavey K, Cortes LS. X-Y translocations and sex differentiation. Semin Reprod Med. 2001;19(2):133-9.
18. Copelli SB, Bregada C, Billerbeck AE, Goldberg AC. Molecular analysis of the Sex Determination in Sex-reversed and True Hermaphroditism. Brazilian J Med Biol Research. 1996; 29(6):743-8.
19. Slaney SF, Chalmers IJ, Affara NA, Chitty LS. An autosomal or X linked mutation results in true hermaphrodites and 46 XX males in the same family. J Med Genet. 1998;35(1):17-22.
20. Meyer J, Sudbeck P, Held M, Wagner T. Mutational analysis of the SOX9 gene in campomelic dysplasia and autosomal sex reversal: lack of genotype/phenotype correlations. Hum Mol Genet. 1997;6(1):91-8.
Recibido: 20 de enero de 2003
Aceptado: 15 de marzo de 2004
Dr. Juan Carlos Piña Napal. Especialista I Grado en Bioquímica Clínica. Master en Genética Médica. Hospital Pediátrico Provincial Docente de Camagüey. Departamento Provincial de Genética Médica. Email: jcpn@shine.cmw.sld.cu

