










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Archivo Médico de Camagüey
versión On-line ISSN 1025-0255
AMC vol.9 no.2 Camagüey mar.-abr. 2005
CASOS CLÍNICOS
Incontinencia pigmenti en el neonato. Informe de un caso
Pigmenti incontinentia in the newborn infant. Case report
Dra. Ingrid Estévez Sierra; Dra. Olga L. Oropesa Vergara; Dra. Alexis Sanchén Casas
Hospital Provincial Ginecobstétrico Ana Betancourt de Mora. Camagüey, Cuba.
RESUMEN
La incontinencia pigmenti (IP) o síndrome de Bloch Zulzberger es una enfermedad rara ligada al cromosoma X, que afecta la piel, pelos, uñas, ojos y el sistema nervioso central. En Cuba no existe diagnóstico de las mutaciones específicas. Ante la rareza de la IP en este medio y la importancia de su conocimiento en neonatos, se presenta el caso de una neonata, natural de Sierra de Cubitas, nacida el 25 de mayo de 2003; 3120 g de peso, parto eutócico. Fue trasladada a la sala de Neonatología del Hospital Materno Provincial Ana Betancourt de Mora porque a las 12 h de vida presentó un cuadro dermatológico caracterizado por lesiones lineales vesiculosas y pustulosas en hemicuerpo derecho. Se planteó el diagnóstico de incontinencia pigmenti. Fue valorada por el genetista y se consideró como caso único y sin antecedentes familiares de esta enfermedad. Se realizaron pruebas de laboratorio clínico, citológico y ultrasonido diagnóstico. Fue indicado tratamiento sistémico y tópico. Fue dada de alta a los 12 días a partir de su nacimiento con evolución de mejorado y seguimiento posterior en consulta de Dermatología.
DeCS: INCONTINENCIA PIGMENTARIA; RECIÉN NACIDO; INFORME DE CASO.
ABSTRACT
Pigmenti incontinentia syndrome of Bloch Zulzberger, is a rare disease linked to X chromosome, which affects the skin, hair, nails, eyes and Central Nervous System. In Cuba there is not diagnosis of specific mutations. Due to the fact that PI is very uncommon in this environment and the importance of its knowledge in newborn infants, all feminine case, from Sierra de Cubitas municipality, born on May 25th, 2003 with 3120 g of weight, eutocic delivery is presented. She was moved to the Neonatology Ward at “Ana Betancourt de Mora” Provincial Maternity Hospital, because at 12 h of life she presented a dermatologic picture characterized by vesiculose lineal and postulous lesions in right hemibody. It was stated the diagnosis of pigmenti incontinentia. She was evaluated by the genetists and considered a unique case without familial antecedents of this disease. Test of clinical, cytology laboratory and diagnostic ultrasound were performed. It was indicated systemic and topic treatment. She was discharged at the 12 d after her birth with improved evolution and posterior follow-up in the Dermatology consultation.
DeCS: PIGMENTI INCONTINENTIA; NEWBORN INFANT; CASE REPORT.
INTRODUCCIÓN
La incontinencia pigmenti (IP) o síndrome de Bloch Zulzberger es un raro trastorno multisistémico y hereditario, transmitido de forma dominante ligado a una mutación del cromosoma X (del par 23), esto ocasiona que el pigmento de la piel, denominado melanina, no se localice en su lugar habitual (en la capa superficial) y se derrame hacia la dermis (capa de la piel que se encuentra por debajo de la epidermis). El gen mutado ubicado en el cromosoma X es dominante. La teoría actual considera esta enfermedad como una displasia neuroectodérmica. Es letal en el varón; la escasez de afectados masculinos y la alta frecuencia de abortos espontáneos apoyan esta suposición. La edad de comienzo es muy variable, puede manifestarse ya desde el nacimiento. 1-4
Este padecimiento afecta fundamentalmente la piel, pelo (alopecia), dientes (retraso y malformaciones), ojos y sistema nervioso central. Por ello, pueden asociarse síntomas como estrabismo, disminución de la agudeza visual, coloboma del globo ocular (fallo en la formación de los órganos oculares). 1-3, 5
Las alteraciones del sistema nervioso central son variables: crecimiento excesivo del tejido cerebral, macrocefalia, dilatación de los ventrículos cerebrales, retraso psicomotor, retraso mental y epilepsia. 3, 4, 6
El criterio diagnóstico más importante es la aparición de un sarpullido progresivo en la piel. Esta lesión atraviesa por cuatro etapas que pueden saltarse y cuya duración es diferente en cada caso: la primera fase es similar a la varicela y puede aparecer al nacimiento o poco después; afecta tronco y extremidades. Posteriormente pasan a una fase verrugosa y luego costrosa y quedan manchas oscuras lineales en forma de remolino. Finalmente, aparece la atrofia en las áreas de piel donde se encontraban las lesiones previas. Por lo tanto, existen dos formas clínicas de presentación: 2, 3, 7
1. En la que sólo se detectan anomalías pigmentarias de la piel.
2. Con afectación sistémica, neurológica y del tracto gastrointestinal.
Es por ello que los pacientes con IP deben ser atendidos por un equipo multidisciplinario constituido además del neonatólogo por un dermatólogo, un estomatólogo, un oftalmólogo, un neurólogo y un genetista.
La literatura internacional refleja un informe de los casos reportados durante 10 años en los Hospitales Infantiles Virgen del Rocío, de Sevilla, y Juan Ramón Jiménez, de Huelva, 3 en seis niñas con edades comprendidas entre un mes y seis años, en las que se diagnosticó IP con un cuadro florido de lesiones cutáneas típicas acompañadas de trastornos oculares, neurológicos, oftalmológicos y dentarios. Por tal motivo y ante la rareza de la IP en este medio, se presenta este caso.
PRESENTACIÓN DEL CASO
Paciente: IPS Historia clínica: 455858
Dirección: Martí No. 12, Sola, Municipio Sierra de Cubitas, Camagüey.
Sexo: Femenino. Raza: Blanca.
Fecha de nacimiento: 23/05/03. Hora: 3:04 am
Parto eutócico: Apgar 9-9 Peso: 3120 g
Ingresado en Servicio cerrado de Neonatología, Hospital Materno Provincial Docente Ana Betancourt de Mora, Camagüey.
Recién nacida, que a las 12 h de vida presentó lesiones eritematovesiculosas en miembros inferiores, por lo que le fue indicado tratamiento tópico con Gentamicina en crema por el neonatólogo.
A las 48 h de nacida empeoraron las lesiones de piel, se tornaron lineales y pustulosas en miembros superiores e inferiores (Fig.1 y Fig.2).
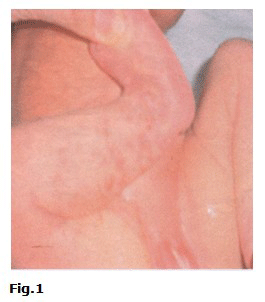
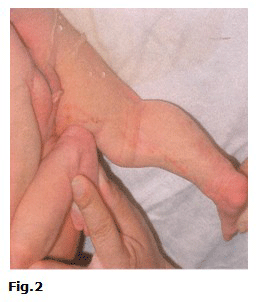
Fue trasladada al servicio de neonatología del propio hospital, para iniciar tratamiento sistémico. Se indicó estudio bacteriológico de las lesiones de piel (positivo a Staphylococcus aureus) y citológico en busca de eosinófilos. Se realizó interconsulta con Dermatología que describió lesiones cutáneas eritematovesiculocostrosas de aspecto abigarrado en miembros inferiores, glúteos y brazos, así como lesiones lineales hiperpigmentadas y verrugosas que se extendían desde el tobillo hasta la región inguinal de la pierna derecha y desde la muñeca hasta la axila del brazo del mismo lado. Se diagnosticó incontinencia pigmenti infectada secundariamente. La citología de las lesiones fueron negativas de eosinófilos. Se realizó examen ultrasonográfico de cráneo, corazón y abdomen, los cuales fueron normales. Se valoró con el genetista, quien concluyó que se trataba de una recién nacida con cuadro dermatológico típico que recordaba al fenotipo clínico de una incontinencia pigmenti, enfermedad ligada al cromosoma X dominante. Se consideró caso único sin antecedentes hereditarios familiares de la enfermedad.
Llevó tratamiento sistémico por 7 d (desde el 25/05/03 hasta el 01/06/03) que consistió en:
Penicilina Cristalina 100 000 UI kg/d y Gentamicina (10 mg) 5 mg por kg/d.
Tratamiento tópico con Gentamicina y Triamcinolona en crema. El día 01/06/03 se suspendió el tratamiento y el 01/06/03 fue dada de alta con seguimiento por Dermatología (Fig.3).
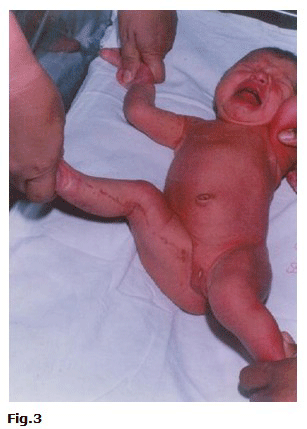
DISCUSIÓN
Por los hallazgos obtenidos en el examen físico y el cuadro clínico de esta neonata, es evidente la presencia de una IP, enfermedad rara, de causa hereditaria, poco reportada a nivel internacional e infrecuente en Cuba. 2, 5, 6
No obstante, se debe considerar el diagnóstico diferencial con otros procesos dermatológicos vesiculares como el herpes simple y el impétigo bulloso o mastocitosis, pero la configuración lineal es única en la IP y el frotis del líquido de la vesícula preparado con la tinción de Wright demuestra masas de eosinófilo. 1-3 Aunque la recién nacida estudiada no presentaba eosinofilia en la citología de las lesiones, es conveniente señalar que se pueden alcanzar cifras hasta de 65 %, esta desaparece a los cuatro o cinco meses de edad.
La base alérgica o autoinmune de este proceso fue comprobada cuando se encontró una eosinofilia en las lesiones vesículobullosas de la piel, en la dermis subyacente, pero estudios posteriores de Jemes y Kurczyski con inmunoglobulinas han desechado esta teoría. 3
A pesar de que no se ha podido involucrar ningún agente infeccioso específico en la evolución de las lesiones de IP, lo cierto es que todos los estadíos descritos en las lesiones dermatológicas se asemejan a otras infecciones virales de la piel como es el herpes simple. 3 Se ha considerado la posibilidad de que en la IP se produzca una disfunción congénita del sistema nervioso autónomo, cuando se expone a infecciones (como es el caso de la neonata del presente estudio) con aislamiento de S. aureus en piel o estímulos ambientales, responsables de las erupciones inflamatorias.
La incidencia familiar de los diversos tipos de anomalías en la IP es elevada, con cifras entre 40 y 55, 4 % de familiares portadores de algún signo clínico que tiene relación con la enfermedad. 3, 4 En la valoración genética de la paciente no se constató la presencia de este factor; no obstante, fue aconsejable un cuidadoso reconocimiento de la madre y los cinco hermanos de la recién nacida, lo que facilitaría el hallazgo de dichos signos mínimos sugerentes de IP, más aún considerando que en el caso que se discute tiene cinco hermanos: cuatro varones y una hembra, y cabía la posibilidad de que tanto alguno de los padres o hermanos tuvieran el cromosoma X mutado.
El predominio abrumador de la IP en las hembras induce a pensar que se trata de una enfermedad ligada al cromosoma X, letal en el varón. 8Algunos autores señalan que podría tratarse de un gen autosómico dominante con mayor expresividad en el sexo femenino. 4, 6, 7
Ante la mejoría de la recién nacida con tratamiento antibiótico para la infección secundaria por estafilococo, la evolución de las lesiones de piel, y la ausencia de alteraciones neurológicas u oculares hasta el momento sugirió el diagnóstico de la forma benigna de presentación de la IP.
No han sido constatadas alteraciones neurológicas ni oculares, se continúa la vigilancia clínica ante la posibilidad de aparición de alguna anomalía del sistema nervioso central.
REFERENCIAS BIBLIOGRÁFICAS
1. Behrman ER, Vaughan VC. Tratado de Pediatría de Nelson: enfermedades de la piel. T 2. 9 ed. La Habana: Edición Revolucionaria; 1988.
2. Esterly N, Solomon M. Trastornos congénitos y hereditarios de la piel. En: Schaeffer A, Avery ME, editores. Enfermedades del recién nacido. T 2. 4 ed. La Habana: Editorial Científico-Técnica; 1981.
3. Rufo M, Sierra A. Facomatosis con discromias extensas: incontinencia pigmenti, nevus acrómico. Rev Neu Barcelona. 1996;24(133):1060-7.
4. Shastry BS. Recent progress in the genetics of incontinentia pigmenti (Bloch-Sulzberger Syndrome). J Hum Gen. 2000;45(6):323-6.
5. Catalano RA. Incontinentia pigmenti. Am J Ophthamol. 1990;110(6):696-700.
6. Landy SJ, Donnai D. Incontinentia pigmenti (Bloch-Sulzberger syndrome). J Med Genet. 1993;30(1):53-9.
7. Francis JS, Sybert VP. Incontinentia pigmenti. Sem Cutan Med Surg. 1997;16(1):54-60.
8. Scheverle AE. Male case of incontinentia pigmenti: case report and review. Am J Med Gen. 1998;77(3):201-18.
Recibido: 28 de octubre de 2003.
Aceptado: 10 de marzo de 2004.
Dra. Ingrid Estévez Sierra Especialista de I Grado en Neonatología. Hospital Provincial Ginecobstétrico Ana Betancourt de Mora. Camagüey, Cuba.

