










 Servicios personalizados
Servicios personalizados Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Coronaviridae family has resurfaced as a result of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic.1) It has been shown that a minor alteration in the SARS-CoV-2 genome will cause a significant change in the structure of drug target proteins, rendering some currently available drugs ineffective.2 As a result, in this current pandemic, it is critical to monitor SARS-CoV-2 genetic diversity. Mutations in the viral genome and functional proteins that help in the virus's adaptation to a new host would inevitably result in such a case of virus dissemination.3 Many attempts have been made to combat the SARS-CoV-2 infection and its global expansion. Natural selection works on variants, and these variations also result in novelty.4 The ability of the virus to evolve and become more compatible with the host may be determined by the age and gender of those infected, as well as the length of time it takes to recover from the disease.5 The nature and frequency of virus mutations remain under study.6 The spike (S), membrane (M), and envelope (E) proteins are essential SARS-CoV-2 proteins that aid in the infection and propagation of the virus within the host cell.7
The E protein is a minor component of the virus membrane that plays a role in virus replication and infection. Coronaviruses without the E protein are promising vaccine candidates because this protein is active in essential aspects of the viral life cycle.8 A hydrophilic N-terminal domain (NTD 7-12 aa), a broad hydrophobic transmembrane domain (25 aa), and a hydrophilic C-terminal domain (CTD) make up the secondary structure of E protein.9
The current study focuses on the E protein because this protein regulates the maturation and retention of the spike protein. We used the available database of SARS-CoV-2 proteins to focus on studying of various mutations in proteins from different isolates in Iraq. Knowing the mutations in SARS-CoV-2's proteins may help to solve the mystery of higher COVID-19 transmission rates that led to the pandemic, as well as provide a helping hand in targeting the virus directly.
Material and Methods
All protein sequences were obtained from the GISAID database. The accession numbers of sequences were taken from NCBI. For databank searches, the FASTA and Blast suites were used.
Multiple sequence alignment and phylogenetic analysis
The selected E protein sequences were QRW43501, QQW45571, QQZ48540, QTP37604, QPI19600, QPI19588, QNL36156, QNL36168, QNL36180, QNL36192, QTH36182, QTH36181, QTH36179, and QTH36176. For alignment, YP_009724392 was selected as a reference wild type. Multiple sequence analysis (MSA) was performed after blasting using CLUSTSAL-OMEGA in EMBL-EBI and MAFFT 7. Results were analyzed and viewed using Jalview.
Prediction of the 2D and 3D structure of E protein
The 3D model of the mutant E protein was constructed using Swiss-Model and Maestro-Schrodinger servers and a homology simulation approach. The PDB (2MM4) comparison of E protein was chosen as a template. TMHMM, DISOPRED, and MEMSAT-SVM were carried out to estimate the two-dimensional structure. ProMod3 is used to build models based on target-template alignment. BioLuminate 4.2 and QMEAN were performed to measure wild and mutant E protein for structural validity and model accuracy (protein processing, covalent bond geometry, protein minimization, residues scanning measurement, energy determination, hydrogen bond optimization, and whole atomic contact analysis). PyMol was applied to display the images. The Schrodinger server was used to calculate the energy of the atoms. Maestro-Schrodinger was used to obtain schematic plots.
Molecular docking
Protein minimization was accomplished using Deploy-YASARA program before starting the molecular docking procedure for E protein (PDB -protein data bank- ID: 7K3G). Molecular docking was carried out using an antiviral agent and antibiotic that may affect the virion particle. The majority of natural ligands interact with E protein in some way. However, the ligands with the lowest dock energy against a specific protein were designated as prospective targets, and their interactions were investigated in depth. We selected doxycycline C22H24N2O8 (ID: 54671203) and rutin C27H30O16 (ID: 5280805) from the PubChem as the perfect ligands of E protein.10 FDA was approved these drugs for healing skin of patients with COVID-19.11 The docking method was carried out utilizing BIOVIA discovery studio and PyRx virtual screening tools for the wild and mutant sequences of the pentameric E protein (7K3G). The Open Babel server was used to minimize the structure of ligands. Vina wizard was used to accomplishing the autodocking.
Results
MSA of 14 E proteins were compared with the wild type (Fig. 1). The findings revealed that the preservation rate for E protein was 98.66 percent due to the appearance of a new mutation (N15Y) in four of the 14 strains studied. In this mutation, the amino acid asparagine is substituted by tyrosine.

When building the 3D structure, PDB (2MM4) was chosen for the E protein. The best recognition scores between template and proteins under analysis were 93%. On the template chosen for the E protein, we constructed a 3D structure of the mutant type. The proportion of modifications in the protein structure and the morphological changes associated with each mutation are depicted in the cartoon and ribbon surface models. The study's results were used to calculate the distance between wild and mutant atoms. The current study revealed that the distance between atoms differs. The distances between the atoms around residue 15 in the wild and mutant strains are different (6.6 Å, 7.6 Å, 3.0 Å, 4.0 Å, and 5.6 Å), (5.9 Å, 5.7 Å, 4.5 Å, 6.2 Å, and 6.1 Å), respectively. The main residue and the whole protein have a less pronounced difference (35.9 Å, 30.3 Å, and 37.5 Å), (35.9 Å, 30.3 Å, and 36.5 Å), respectively.
A pentamer structure of E protein (PDB ID: 7K3G) consists of five chains. The mutation was first loaded onto one chain, then onto all links. By homology modeling and constructing the topology structure, we established that there is a difference in how a mutation affects the overall shape of the protein, especially if the mutation occurs in all chains. The new protein has a completely different topology from the old one, which has an effect on the protein and, of course, the virion particle (Fig. 2).

Fig. 2 Prediction of the pentamer E protein (7K3G) and constructing mutation. A: Wild type 1- Cartoon with N15 residue in all chains, red color. 2-Surface side view. 3- Cross-section. 4- Distances between chains of atoms N15 residue. B- One chain mutation of Y15. 1- Cartoon with Y15. 2- Surface side view. 3- Cross-section shows a pocket in Y15 residue. 4- Distances between chains of atoms Y15 residue. C: All chains mutated with Y15. 1- Cartoon with Y15 in all chains, red color. 2- Surface side view shows a large hole. 3- Cross-section presents 2 pockets. 4- Distance between chains of atoms Y15 residue.
Before beginning the molecular fusion process, the E protein (7K3G) was miniaturized, yielding the following: the initial energy is (-47628.5kJ/mol) with a score of (-5.29) and the final energy is (-71100.2 kJ/mol) with a score of (-1.77). According to the docking results between E protein and ligands, the molecular docking of nine models of each type of ligand has a high value when compared to the wild and mutant types. When the root-mean-square deviation (RMSD) is zero, the optimal degree of coalescence energy has been chosen. Model number 1 was selected for each kind because it had the maximum degree of fusion, as indicated in Table 1. The binding affinity of the wild type was greater than the mutant type for rutin: -8.6 kcal/mol, -8.3 kcal/mol respectively. This study found that the mutant type's binding energy for the doxycycline ligand is higher than the wild type's: -8.3kcal/mol and -6.8kcal/mol, respectively.
Table 1 Binding affinity scores of molecular docking models (protein-ligand interaction) and the degree of RMSD for both rutin and doxycycline ligands.
| Wild type-Ligand | Binding Affinity | RMSD/ub | RMSD/lb | Mutant type-Ligand | Binding Affinity | RMSD/ub | RMSD/lb |
|---|---|---|---|---|---|---|---|
| 7k3g-Rutin Model 1* | -8.6 | 0 | 0 | 7k3gM-Rutin Model 1 | -8.3 | 0 | 0 |
| 7k3g-Rutin Model 2 | -8.5 | 3.925 | 1.723 | 7k3gM-Rutin Model 2 | -8.1 | 4.799 | 2.071 |
| 7k3g-Rutin Model 3 | -8.5 | 2.416 | 1.682 | 7k3gM-Rutin Model 3 | -7.9 | 7.403 | 2.095 |
| 7k3g-Rutin Model 4 | -8.5 | 2.425 | 1.69 | 7k3gM-Rutin Model 4 | -7.8 | 4.242 | 2.255 |
| 7k3g-Rutin Model 5 | -8.2 | 15.214 | 13.009 | 7k3gM-Rutin Model 5 | -7.6 | 4.327 | 1.818 |
| 7k3g-Rutin Model 6 | -8.2 | 15.927 | 13.441 | 7k3gM-Rutin Model 6 | -7.6 | 6.98 | 2.419 |
| 7k3g-Rutin Model 7 | -8.2 | 15.504 | 13.207 | 7k3gM-Rutin Model 7 | -7.5 | 5.569 | 2.639 |
| 7k3g-Rutin Model 8 | -8.2 | 15.622 | 13.196 | 7k3gM-Rutin Model 8 | -7.5 | 7.021 | 1.87 |
| 7k3g-Rutin Model 9 | -8.2 | 15.88 | 13.453 | 7k3gM-Rutin Model 9 | -7.5 | 7.191 | 2.465 |
| 7k3g-Doxycycline Model 1** | -6.8 | 0 | 0 | 7k3gM-Doxycycline Model 1 | -8.3 | 0 | 0 |
| 7k3g-Doxycycline Model 2 | -6.8 | 6.532 | 3.718 | 7k3gM-Doxycycline Model 2 | -7.8 | 1.787 | 1.543 |
| 7k3g-Doxycycline Model 3 | -6.8 | 4.037 | 2.462 | 7k3gM-Doxycycline Model 3 | -7.4 | 5.785 | 3.029 |
| 7k3g-Doxycycline Model 4 | -6.8 | 4.035 | 2.448 | 7k3gM-Doxycycline Model 4 | -7.3 | 7.104 | 2.251 |
| 7k3g-Doxycycline Model 5 | -6.7 | 6.404 | 3.516 | 7k3gM-Doxycycline Model 5 | -7 | 6.961 | 3.102 |
| 7k3g-Doxycycline Model 6 | -6.7 | 4.885 | 3.116 | 7k3gM-Doxycycline Model 6 | -7 | 6.375 | 2.308 |
| 7k3g-Doxycycline Model 7 | -6.7 | 6.05 | 2.827 | 7k3gM-Doxycycline Model 7 | -6.9 | 3.586 | 2.208 |
| 7k3g-Doxycycline Model 8 | -6.7 | 5.454 | 3.742 | 7k3gM-Doxycycline Model 8 | -6.8 | 2.024 | 1.509 |
| 7k3g-Doxycycline Model 9 | -6.3 | 38.61 | 36.046 | 7k3gM-Doxycycline Model 9 | -6.7 | 6.259 | 2.867 |
Model 1*: the highest score for 7K3G-Rutin for wild and mutant type. Model 1**: the highest score for 7K3G-Doxycycline for wild and mutant type.
According to the 2D structure of (7K3G-Doxycycline), the main interacting residues in the E protein are GLU8, THR11, and ASN15 for the wild type. The resultant mutant type (7K3GM-Doxycycline), in which the amino acid asparagine was replaced with tyrosine, shows a shift in the degree of contact. It possesses an unstable bond link, but one with greater energy than in the wild type, and the fusion now encompasses the residues GLU8, THR11, and TYR15, as seen in Figure 3. VAL25, LEU28, and ALA32 are the major interacting residues in the wild type (7K3G-Rutin). On the other hand, the interaction between residues of mutant strain (7K3GM-Rutin) resulted in a significant alteration in the docking site. The interacting residues (GLU8, THR11, and TYR15) share the same interaction with a mutant type of the doxycycline ligand (Fig. 4). This finding suggests that the new mutation in the E protein has altered the degree of binding and impacted the ligand-binding site.

Fig. 3 2D structure of molecular docking interaction residues showing the types of hydrogen, conventional and unfavorable bonds of the wild strain 7K3G-Doxycycline and mutant strain 7K3GM-Doxycycline.
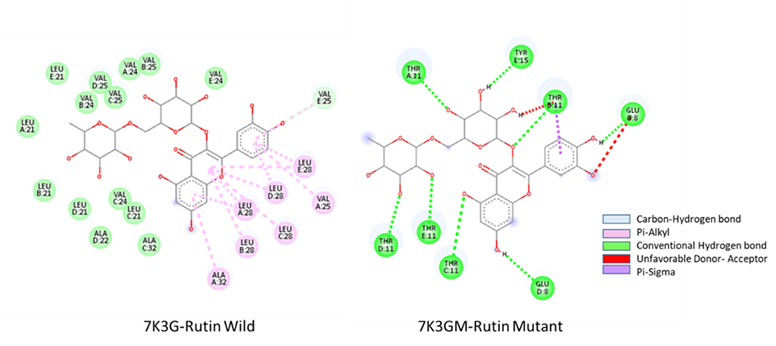
Fig. 4 2D structure of molecular docking interaction residues showing the types of hydrogen, Pi-Alkyi, unfavorable donor and acceptor, and Pi-Sigma bonds of the wild strain 7K3G-Rutin and mutant strain 7K3GM-Rutin.
We detected a conformational change in the shape of the mutant protein, in relation to the wild type, after constructing the 3D structure. In addition, as shown in Figure 5, the degree of conferring hydrogen bonding was greater for the wild type of doxycycline docking. In the case of the ligand rutin, it was registered that a mutation in position N15Y changed the structure of the protein dramatically. In addition to altering the docking location, the mutation residue became part of the docking site, as seen in Figure 6.

Fig. 5 Molecular docking of the E protein and doxycycline ligand. A: Wild E protein surface shows ASN15 in red color reacting with the doxycycline in green ribbon stick. B: Mutant E protein surface shows TYR15 in red color reacting with doxycycline in green ribbon stick. C: Wild docking residue shows the degree of difference between the donor and the acceptor of the hydrogen bonds. D: Mutant docking residue shows the degree of difference between the donor and the acceptor of the hydrogen bonds.
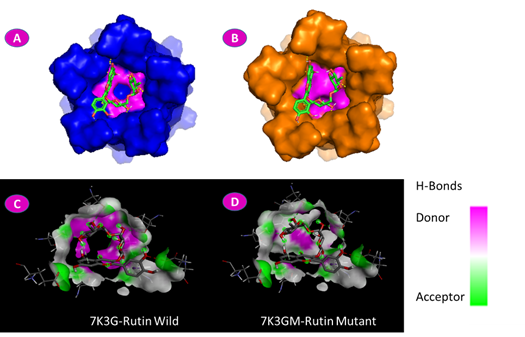
Fig. 6 Molecular docking of the E protein and rutin ligand. A: Wild E protein surface shows ASN15 in magenta color reacting with the rutin in green ribbon stick. B: Mutant E protein surface shows TYR15 in magenta color reacting with rutin in green ribbon stick. C: Wild docking residue shows the degree of difference between the donor and the acceptor of the hydrogen bonds. D: Mutant docking residue.
Discussion
According to popular belief, the latest COVID-19 pandemic was caused by the cross-species transmission of alpha-coronavirus, commonly found in bats and possibly pangolins, to humans. E protein has been linked to viral entry, replication, and particle assembly in human cells.11 Understanding the development of this modern coronavirus and assuring the performance of new diagnostic tests, vaccinations, and therapies against COVID-19 requires tracking SARS-CoV-2 genetic variation and evolving mutations in this continuing pandemic. The amino acid heterogeneity of the SARS-CoV-2 E and M structural proteins is investigated in this descriptive analysis.12
This study detected a mutation in the E protein that significantly impacted the protein's conformational transition. The total percentage of mutation occurring in our current research does not align with previous studies. For example, a study revealed that 99.99% of the 103,419 E sequences studied were conserved.13 Furthermore, E protein had fewer amino acid modifications, despite having almost equal retention 99.98%.14 The mutation rate was less than 1% in all geographic regions except Africa, where 92% of the epiweeks with African sequences available belonged to the most recent epiweeks. According to epiweek's review, there was no consistent growth over time, either nationally or regionally.15,16
A short (7-12 aa) hydrophilic N-terminal domain (NTD), a broad hydrophobic transmembrane domain (25 aa) with a high proportion of valine and leucine, and a hydrophilic C-terminal domain (CTD) make up the coronaviruses E secondary structure.17 Despite the mutation location changed from helices to coils, these characteristics were found in the secondary structure of the E protein. Consequently, the frequency of residues was altered in the wild type compared to the mutagenic variant.
The structure of the E protein changed dramatically as a result of the mutation. Our results confirm that the rate of mutant variants is 6% from all sequences isolated in Iraq. This demonstrates that the measurements vary between the atoms at the mutation residue and the protein tip site. Furthermore, the amount of energy measured for the atoms at different levels was low for covalent bonds and strong for solvated bonds. The protein becomes more stable as a result of this mutation.
In terms of the E protein, the pentamer model (7K3G) was selected to confirm the impact of the single mutation on the protein's overall form. For a single chain and all chains, the 3D structure of the protein in the mutation state was built. The existence of a new pocket appeared within the protein in the case of a single chain mutation. Still, the analysis discovered two internal pockets as a result of the consequence of the combined five chains mutation. We assume that this effect would impact the virion's overall shape and effects on the host cell.
According to our findings, the docking of the doxycycline revealed the presence of additional connections in the mutant position and a rise in the degree of fusion. This may call attention to the possibility that doxycycline has a direct effect on the virus by enhancing its binding to the E protein. The effect of the mutation was proven in the case of rutin by altering the ligand-protein binding site, which is also helpful in practice.18
It's worth noting that the mutant type has a lower degree of hydrogen bonding than the wild type for both ligands. We believe the study will continue looking for other ligands that affect the protein, perhaps opening up a new sector in COVID-19 therapy.
Unfortunately, it is impossible to know if the positions of these changes are exposed to the membrane's internal or external hand. In either case, the replacement and deletion are major alterations that may affect conformational properties and potentially protein-protein interactions. More systemic research is needed. However, these modifications may affect the oligomerization mechanism that leads to the formation of a transmembrane ion channel.19,20
Understanding amino acid modifications´ global and regional effects would require further research into related mutations, virus-host protein interactions, and protein structure. Comparative researches may, therefore, shed light on the molecular processes underlying the emergence of an outbreak of epizootic origin, as well as propose molecular targets for therapeutics or reverse vaccinology experiments.
Conclusions
This study investigated a significant mutation in the E protein that directly impacted the protein's shape and, as a result, on the virion particle as a whole. The mutation altered the degree of binding for doxycycline by directly affecting the ligands' attachment residues. The mutation altered the location of the rutin's attachment to the E protein, which has an impact on the virion particle.
Acknowledgment
This research was funded by the vice-chancellor for research of Jahrom University of Medical Sciences and Health Services. The authors are appreciative to all the patients and their families for their kind cooperation in this research.

