










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBiotecnología Aplicada
versión On-line ISSN 1027-2852
Biotecnol Apl vol.31 no.4 La Habana oct.-dic. 2014
RESEARCH
Collection of Salmonella typhimurium strains for genotoxicologic and anti-genotoxicologic evaluation
Colección de cepas de Salmonella typhimurium para la evaluación toxicológica y anti-genotoxicológica
Elizabeth B Cuétara1, Angel Sánchez-Lamar2, Blanca E Hernández-Guadarrama3, Javier J Espinosa-Aguirre3, Rafael Camacho-Carranza3
1 Department of Clinical Experimental Pharmacology, Research Division,National Institute of Oncology and Radiobiology, INOR. Calle 29 y F, Vedado, CP 10 400, La Habana, Cuba.
2 Genetic Toxicology Laboratory, Faculty of Biology, University of Havana. Calle 25, No. 455 e/ I y J, Vedado, CP 10400, La Habana, Cuba.
3 Departamento de Medicina Genómica y Toxicología Ambiental, Instituto de Investigaciones Biomédicas, Universidad Nacional Autónoma de México. Tercer Circuito Cultural Mtro. Mario de la Cueva, Ciudad Universitaria, Apartado Postal 70228, México, D. F. 04510, México.
ABSTRACT
Homologous recombination is a DNA repair mechanism that additionally generates a biological diversity. Most of our knowledge about it was obtained from assays that used double strand ends as initiators of recombination. The present work is aimed at evaluating homologous recombination in bacteria, using an assay based on the segregation of chromosomal duplication. This assay measures exchanges between sister strands without favoring any pathway. A collection of strains of Salmonella enterica serovar typhimurium deficient in genes coding for proteins involved in homologous recombination was built. We evaluated spontaneous (SSR) and UV-induced segregation rate (UV-ISR). We demonstrated that the absence of RuvC resolvase did not affect SSR, while defects in RecA, RecQ and RecB/RecF decreased it and the lack of SbcCD or RuvAB stimulated it. In RecB null mutants, lesions that are not commonly sensed by RecFOR seem to have been recognized and repaired by this pathway. The methodology supported the corroboration of a RecA-independent pathway in Salmonella and suggests the existence of alternative recombination pathways in recB/recF double mutants.
Keywords: homologous recombination, DNA damage, UV light, bacteria, Salmonella.
RESUMEN
La recombinación homóloga es un mecanismo de reparación del ADN que adicionalmente genera diversidad biológica. Casi todo nuestro conocimiento acerca de ella proviene de ensayos que emplean extremos de doble cadena como iniciadores de recombinación. En este trabajo se evaluó la recombinación homóloga en bacterias, mediante un ensayo de segregación de duplicaciones cromosomales que mide los intercambios entre cadenas de cromátidas hermanas sin favorecer ninguna ruta de reparación. Se construyó una colección de cepas de Salmonella enterica serovar typhimurium deficiente en genes que codifican proteínas involucradas en la recombinación homóloga. Se evaluó las tasas de segregación espontáneas (SSR) e inducidas por luz ultravioleta (UV-ISR). Se demostró que la ausencia de la resolvasa RuvC no afecta la SSR, mientras que los defectos en RecA, RecQ y RecB/RecF la disminuyen y la carencia de SbcCD o RuvAB la estimulan. En mutantes nulos para RecB, las lesiones que usualmente no son detectadas por el sistema RecFOR parecen ser reconocidas y reparadas por esta vía. La metodología usada permitió corroborar la existencia de una ruta independiente de RecA en Salmonella y sugiere la existencia de rutas alternativas de recombinación para el doble mutante recB/recF.
Palabras clave: recombinación homóloga, daño al ADN, luz ultravioleta, bacterias, Salmonella.
INTRODUCTION
The maintenance and propagation of the genetic material is a prerequisite for life. Environmental hazards constantly challenge chromosomal integrity of the cells [1]. DNA is constantly damaged by both endogenous and exogenous sources, and genotoxicity can be considered as an imbalance between DNA damage and repair mechanisms [2]. Solar ultraviolet (UV) irradiation is widely known as a genotoxic environmental factor. The increasing UV incidence levels that have been affecting both, the biosphere and humans, is the main consequence of the ozone layer reduction of the last decades. That is why scientists have been making many efforts to understand the role of sunlight in the induction of DNA damage, mutagenesis, and cell death [3, 4].
Ultraviolet radiation can kill cells and transform the properties of survivors. Because radiation produces both effects, it is reasonable that they might be related [5]. It is widely accepted that nucleotide excision repair (NER) pathway plays an outstanding role in UV-induced DNA lesions repair [6]. Nevertheless, since the 1970’s there was suggested that recombination mechanisms complement excision mechanisms in UV-induced damage repair [7]. So, UV light is not a direct inductor of homologous recombination (HR) but the processing of UV-induced damage generates recombination initiators.
HR was originally described as the result of the sexual process but later it was identified as a major DNA repair process. It is widely accepted that in bacteria, the primary function of HR is the repairing of collapsed replication forks, which is a surprisingly common process, even under normal growth conditions [8]. This kind of repair can also be triggered by the presence of its substrates: double strand breaks (DSB) and single strand gaps (SSG). Recombination assays that use DSB as initiator, commonly overestimate the importance of RecBCD in HR, regarding RecF as an accessory pathways. Thus, we decided to use a duplication-segregation assay (dup-seg assay) [9] for assessing recombination processes induced by a non-classical initiator of HR, cyclobutyl pyrimidine dimers (CPDs), to reveal the participation of some proteins of the two described pathways: RecBCD and RecFOR, without favoring any of them. Segregants scored arise mainly by a single intramolecular non-reciprocal exchange between direct chromosomal repeats. In this assay, the direct-repeats flanking markers confer lactose utilization ability and ampicillin resistance (Lac+, AmpR). Homologous recombination events generate Lac deficient (Lac-) ampicillin sensible (AmpS) segregant phenotypes that can be visualized by culturing the cells in a nutrient broth (NB) medium supplemented with X-gal (a β-galactosidase substrate). Recombination was evaluated as segregation rates in a wild type strain and strains deficient in several proteins involved in different steps of homologous recombination [10].
MATERIALS AND METHODS
Salmonella typhimurium strains
We used a wild type strain (a base strain with a duplication of 36.2 kb, ampicillin resistance and Lac proficiency) and eight strains mutated in genes involved in the known pathways of HR, RecBCD and RecFOR. Such strains were obtained by phage transduction in the base strain (Table 1).
Cell culture
Cells were cultured in NB medium (8 g/L, Difco Laboratories) with NaCl (5 g/L). Solid medium contained Agar Base (40 g/L, Difco). Plates used to score bacterial phenotype contained the 5-bromo-4-chloro-3-indolyl-γ-D-galactopyranoside (X-gal) chromogenic β-galactosidase substrate at a final concentration of 40 μg/mL. The substrate was obtained from Sigma Chemical Co. and antibiotics from Sigma Chemical, Co. Final concentrations of antibiotics were: 30 μg/ mL sodium ampicillin (Ap), for single-copy elements (MudA); 20 μg/mL chloramphenicol (Cm); 20 μg/ mL tetracycline hydrochloride (Tc); 50 μg/mL kanamycin sulfate (Km) (Table 1). All incubations were performed at 37 ºC and 100 rpm of agitation.
Duplication-segregation assay
The assay was performed as described by Espinosa- Aguirre et al. [10]. Briefly, 200 μL of an overnight culture was inoculated in 10 mL NB medium, plus the corresponding antibiotics and it was allowed to grow to exponential phase (OD600 nm = 0.4). It was then diluted two times; 1:10 000 in saline solution and 1:10 in fresh NB suppressing antibiotic restriction to reach a final concentration of 1000 cells/mL. This sample was divided into two portions: one to determine SSR and the other for mutagen treatment to evaluate UV-ISR. At this moment, two plates per treatment were cultured (10 and 100 μL) to testify that there were no white colonies, allowing the experiment to be continued. Irradiation was performed as described below. Then, 100 μL (approximately 50 cells) were placed in 96-well plates for 16 hours at 37 ºC. Twenty independent cultures were grown per treatment. After incubation, cells were again diluted 1:1 000 000, and then 10 and 100 μL of such dilutions were spread in plates containing NB X-gal and further incubated for two days. Colony forming units were visually scored. From the total, white colonies (segregants) were counted. The structure of the chromosome duplication-segregation is shown in Figure 1. Data was used to calculate segregation rate (μ) using the Luria-Delbrück method known as “Fluctuation test” [10, 11]. Statistical significance was considered when values exceeded three times the standard deviation (μ ± 3sμ).
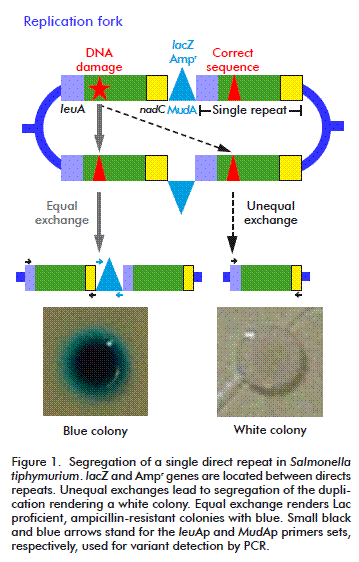
In order to test that segregants were recombinants and not lac mutants, a PCR was run between the right edge of the MudA element and the 5´ region of the leuA fragment joint to the element (~850 bp; 35 cycles, 59.2 ºC annealing; primer set MudAp: forward-5´-GAAACGCTTTCGCGTTTTTCGTGC-3´and reverse- 5´-GCCAGCAAGTCATTATTTTTGATACGACC-3´); after the segregation, those sites must be lost. The intact leuA gen (1721 bp; 35 cycles, 67° C annealing, primer set leuAp: primers forward-CGCCCGGCAGTACGGCAATA reverse-GACCGGTGGGCGGCATTCAA) was amplified as control.
Irradiation with UV light
Samples of 5 mL of an exponential phase culture (cells diluted 1: 10 000 in saline solution and 1: 10 in fresh NB) were placed in a sterile, 5 cm of diameter petri dish. The lid was removed for irradiation once inside the UVP CL-100 UV Ultraviolet Crosslinker equipment (using five Hg black light bulbs F8T5). Two doses were used: 2 and 10 J/cm2, at room temperature.
RESULTS AND DISCUSSION
SSR on mutant strains deficient in HR proteins
Proficient and deficient HR strains were characterized for SSR (Table 2). The lack of a protein involved in a HR pathway did not always decreased spontaneous recombination capacity of the strain. RuvC deficiency did not affect Salmonella’s ability to recombine. When RuvC is not available, there are other proteins (RusA or topoisomerase IV) that could assume its function. RuvC is widely represented in bacteria and it is also known that proteins from one organism can act in other organisms in spite of differences in se-quence. According to such findings, Aravind et al., proposed a RuvAB-independent mechanism of action for RuvC [12]. RusA is another resolvase that could act instead of RuvC, although is it not evolutionarily related to the known resolvases family [13]. RusA is able to join the Holliday junctions (HJ) affecting its global structure [14]. Comparative analysis using agarose gel electrophoresis pointed out to the formation of symmetric structures in tetrameric conformation [15]. In addition, Zechiedrich et al. demonstrated that topoisomerase IV is the most important decatenase in E. coli and S. typhimurium replication and recombination [16]. Hence, we suggest it could replace RuvC activity.

Deficiencies in SbcCD, RuvAB and RecB increased the SSR. SbcCD is a member of the structural maintenance chromosome family. It has been demonstrated that this protein degrades dsDNA in the 3´→5´ direction, releasing a product half the length of the original one [17]. These observations support the idea that the SbcCD enzyme degrades fork structures that inhibit replication and recombination intermediaries, suppressing the illegitimate exchanges by degrading exogenous DNA and using precursors as nutrients [18]. On the other hand, bacteria and eukaryotes carry long palindromic sequences that result in genomic instability sites (deletions, amplifications and translocations). There is evidence that these are SbcCD’s cleavage sites and can be repaired by PriA/ RecBCD-mediated HR, related to replication but not occurring in the fork [19]. In the absence of SbcCD function, the lifetime of its substrates increases and they can be recognized and processed by alternative ways. According to our data, this occurs quickly and efficiently, exceeding the wild type strain capacity. Our results coincide with those by Miesel and Roth [20], who reported that in RecB null strains, RecFOR pathway is capable of initiating HR from a double strand end with a single strand extension, though they also need to mutate SbcB.
It was unnecessary to have the double mutation recB/sbcCD to identify the RecFOR system action. The evaluated recB null strain showed a higher recombination capacity than the wild type, similar to results by Miesel and Roth working with a RecD mutant and a short sequence transduction assay in a Salmonella model [20]. In fact, hyper-recombinogenic recB deficient strains have been described before. In 2011, it was reported that a recB null mutant strain exhibited a constitutive RecJ nuclease-dependent SOS response.
Another strain with a high frequency of recombination is the ruvAB mutant. Bacterial helicases such as: RuvAB, DnaB, RecG and RecQ have shown in vitro branch migration activity [21]. RuvAB and RecG participate in the conversion of the DNA-protein initial complex and HJ resolution. RuvA and RuvB are involved in regression and resolution, respectively. According to our experience, RuvAB deficiency slightly increased the Salmonella’s SSR. The role of RuvAB could be assumed by RecG, which processes Holliday’s intermediaries in vivo. Another option has been suggested by López et al.: RecQ can recognize and unroll DNA structures forming substrates for topoisomerase III in vitro [22]. Additionally, RecQ-mediated branch migration could join two Holliday structures forming substrates released by topo-III. Moreover, it have been demonstrated little or no control in crossing-over assays at the level of HJ and the need for complementary factors to determine the protein cell uses in every situation [23].
The strains mutated in recA, recQ and recB/recF show lower spontaneous recombination frequencies. Some authors pointed out that the genetics of recom-bination is assay-specific [24], making the interpretation of results complicated. Even though, our results are in line with the common view of RecA deficient strains as having low segregation rates which increase when homology size rises, but not in the same magnitude than in RecA-dependent pathways. Fluctuation tests have been used generally to determine μ and to measure the host cells capacity of being transformed by plasmids. Recent reports evidenced the existence of RecA-independent pathways in E. coli, analyzing conversion and crossover events in mutants lacking exonucleases. Such mutants increased their RecA-independent recombination levels to similar proportions of RecA-dependent recombination [25]. This indicated that RecA-independent recombination is not inefficient, but limited by substrates and tending to occur in replication forks.
There are other biological processes that involve RecA-independent recombination. Swingle et al. demonstrated the existence of a highly conserved RecA-independent mechanism for the integration of oligonucleotides in bacterial chromosomes of E. coli, S. typhimurium, Shigella flexneri and Pseudomonas syringae [26]. In recA mutant cells, recombination occurs at low but detectable levels. Besides, systems as λ red recombination stimulate RecA-independent recombination between chromosomes if at least one of them is replicating. When there is not replication, recombination occurs via a RecA-dependent pathway [27].
RecQ deficient strains decreased seven times the spontaneous recombination ability of Salmonella in the dup-seg assay. Our results are opposite to the findings of Ivanovik and Dermic, who used Hfr-conjugation and P1 transduction assays to assess the recombinogenic capacity of E. coli [28]. Those assays implicate double strand ends as HR initiators, thus favoring the RecBCD pathway. In the dup-seg assay, any lesion could trigger recombination, and then, the RecFOR pathway could be activated. RecQ is an element of this pathway and its deficiency can limit the number of recombination events.
The loss of RecF function reduced SSR to the half in bacterial cells used in our experiments. RecF affects the extension of RecA nucleofilament on DNA by interacting physically with RecX (a RecA inhibitor) [29]. Considering it, a possible explanation to μ decreasing could be RecX’s free acting. There are two pathways that involve the RecF, RecO and RecR proteins: the RecFOR and RecOR. The presence of RecF blocks the RecOR pathway and stimulates the activation of RecFOR, which slowly happens if there are no duplex regions near to ssDNA where RecA can be loaded. In the absence of RecF, RecBCD and RecOR pathways are favored, involving the interaction of the RecO with the C-terminal of SSB proteins. Recombination decays indicate the presence of multiple single strain cuts that could be assumed by the duet RecOR acting independently, justifying a not too drastic decrement of recombination [30]. In fact, recF mutations delay SOS induction [31].
The SSR of the double mutant strain was very low (twenty two times lower), similar to that of the recA deficient strain. This was expected due to the respective total and partial impediment of RecBCD and RecFOR. Our results coincide with those reported for a recB1080, recQ, xonA mutant (two pathways limited), of low viability and high levels of illegitimate recombination [28]. Besides, damage could be repaired by inducing translesion synthesis (TLS) polymerases. TLS is error prone and, due to the low fidelity of the enzymes involved, it could be responsible for segregants derived from our double mutant [32].
Other mechanisms of RecBCD and RecFOR-independent recombination have been described. They involve RecA, due to its ability for nucleation and extension on DNA, after the recognition of tetraplex structures in palindromic sequences [33], or the generation of HJ in collapsed replication forks, related with the inactivation of DnaB without inducing SOS or recruitment of pre-synaptic proteins [34].
UV-ISR
UV-ISR results are shown in (Figure 2). Our data evidenced four types of behavior in response to UV treatment, hereafter called types I, II, III and IV.
Behavior type-I
RecF, RuvAB and RuvC deficient strains behaved in the same way than the wild type strain, increasing its mutability after low doses of UV treatment and slightly reducing it at the high UV dose. Cells with mutations in recF were unable to process induced DNA damage and replication restart due to irradiation [35]. At high UV doses, such failure is not drastic because RecQ helicase and RecJ nuclease alternatively degrade the nascent strand until NER or TLS allow replication restart [1]. It was demonstrated that the nature of the replication arrest defines the way of replication restart in UV-induced collapsed forks [36].
On the other hand, photoproducts like CPDs, disrupt hydrogen bridges in adduct vicinity, generating physical stress in supercoiled DNA, formation of cruciform structures or conversion from B to Z form to liberate tensions. These intermediaries resemble the recombination ones and can be substrates for dynamic reactions mediated by the RuvAB complex. Thus, DNA fragmentation depends on active DNA synthesis previous to UV irradiation [37]. It seems that at low UV doses, DNA damage processing depends only on strand exchange mediated by RecA and the resolution of HJ by RuvABC. In our experiments, RuvC-cells behave very similar to RuvAB-. It has been observed by using pulse field gel electrophoresis analysis of Xba I-digested DNA, that ruvC-mutants treated with 5 J/m2 of UV filamented and their chromosomes were aggregated in the central region [38]. Those results were explained as mediated by abortive recombination that keeps together the new chromosomes. Other attempts failed to identify a new resolvase in an E. coli ruvABC- strain (N4237) by mapping and molecular biology techniques, explaining mutant survival throughout RusA expression due to an insertion of a transposon Tn10dkan upstream the rusA gene [39]. In our opinion, RuvAB, RuvC and RecF are dispensable in UV-induced DNA damage because μ varies in their absence as for a wild type strain. It could be explained assuming that RecBCD pathways play the key role of repair and Ruv’s proteins have efficient replacements in Salmonella.
Behavior type-II
Strains deficient in RecA, RecQ and SbcCD exhibited a type II behavior. Their frequency of recombination decreased at low UV doses and increased at high UV doses, overriding SSR in the first cases.
UV-ISR observed in recA and recQ mutants at the 2 J/cm2 dose can be explained by the transient reversion of damaged replication forks, which could be stabilized by RecA and with the participation of proteins associated with the RecFOR pathway (RecQ), delaying the process enough to allow repairing by NER before replication restart. This was demonstrated by Jeiranian et al., using E. coli cells and bi-dimensional electrophoresis in agarose gels [40]. They detected intermediary structures originated in vivo from plasmid DNA after UV light treatment. RecA- mutants failed to restart replication post-irradiation and exhibited a quick and eventually complete degradation of the chromosome [41]. The high number of segregants obtained at a dose five times higher, can be explained by the appearance of clustered lesions. Isolated lesions can be easily and quickly repaired in an efficient way. Nevertheless, it has been suggested that lesions clustering are quite difficult, if not impossible, to repair without HR. Reparability of DNA clustered lesions is determined by the identity of component lesions and the relative distance among them [42]. We may be visualizing the appearance of the mutant phenotype due to deletion and not to proper segregation of the duplication. Further studies will be necessary to clarify the predominant alternative.
Bacterial cells have developed alternative mechanisms to process gaps covered by RecA: HR, TLS and RecA-mediated excision repair (RAMER). The first two have already been commented. The last one comprises the RecA mediated pairing of single stranded DNA with an undamaged homologous, UvrABC-mediated repair during the exchange involving polymerization. The mechanism selected depends on the mediator availability even of post-transductional modification of TLS polymerases (mainly Pol-IV and Pol-V) [43].
On the other hand, RecA appears to be more important for deletions than for duplication formation. Supporting these observations, Reams et al. proposed that the strand exchange activity of RecA contributes equally to the processes of deletion and duplication [44]. Reduced HR caused by the lack of RecA may be compensated by an increase in single-strand annealing. This increase may occur because DNA breaks required for annealing persist longer in the absence RecA, making it more likely for two breaks to coexist. Moreover, revealed single strands may anneal better when not involved in RecA protein filaments.
Suppression of illegitimate recombination, collapse fork restart and SOS induction are affected in recQ mutant cells [45]. They have a diminished capacity of recombination in the presence of non-coding lesions (CPDs) that render recombination intermediaries. Hua et al. demonstrated that RecQ participated in UV-induced damage repair through the interaction with NER proteins in uvrA1, uvrA2 and uvsE mutants [46]. The evaluation of E. coli recQ- mutants evidenced a delay in SOS response. SOS induction is triggered by interaction of activated-RecA with LexA [47], but RecQ is necessary for the fast degradation of the LexA repressor in response to UV injury [48]. They proposed a mechanism that involves RecQ and RecJ. If the UV dose increases then NER could be saturated and RecA-independent mechanisms may act, explaining the observed increment in μ. Our results differ from those of Nakayama et al., who found that mutation in recQ neither affect viability nor conjugation capacity [49]. Other authors have explored the behavior of exonucleases’ mutants (e.g. SbcCD) in a recBCD+ background, exposed to UV light [50], and evidenced that simple or combined deficiency in single strand DNA nucleases increased UV sensibility using transduction and conjugation-based methods. Our results are comparable to those at low doses.
Behavior type-III
The recB mutants’ ability to recombine after UV treatment decreased dramatically with increasing UV doses (type III behavior). Our results agree with those of Feng and Hays [51], showing a reduction of the unwinding capacity and processativity in recombination events using λ phages.
Behavior type-IV
Regarding the recB/recF mutant strain, the reduction of the number of colony forming units associated with an increment of UV-ISR. At high UV doses, corresponding to low survival, the ratio of transformed to surviving cells asymptotically approached an upper limit [5]. This suggests that survival is conditioned by the mutation ability and it is also plausible as related to SOS response. Some recB mutants exhibit constitutive expression of the SOS regulon [52]. The level of constitutive or induced SOS response depends on the timely signaling and repairing capacities. A passive mechanism for RecA loading, independent of RecBCD or RecFOR and mediated by RecJ, have been described [53]. So far, we assume the existence of two pathways for HR: RecBCD and RecFOR, though there is evidence of imbrications among them. Experiences using short homology assays demonstrated that RecF, RecJ and SbcB participate in RecBCD-dependent events [20].
To corroborate in our experiment if segregants (white colonies) were true recombinants or just colonies with punctual mutation in the lac gene, we made a PCR between the right edge of the Mud element and the 5´ region of the leuA fragment joint to the element, the results shown in Figure 3.

Three subsets were identified. The first corresponded to the DNA amplification from colonies with the duplication, the second to segregants and the third to the TR7069 amplification product (wild type). The expected band identifying the fusion with the mud element had an approximated size of 850 bp, been only detected in strains with the duplication and absent in the wild type and segregant strains.
CONCLUDING REMARKS
The dup-seg assay was able to detect the RecFOR pathway events without introducing additional mutations in recBCD deficient background. Our results indicate that RecQ is a key protein and that RecA-independent recombination occurs in S. typhimurium.
Bacterial assays are useful tools for assessing DNA damage and repair, due to the flexibility of the design, its low cost and rapid obtainment of results. They also offer the possibility of data extrapolation to eukaryotes. For example, Casper et al. used a chimerical yeast with a human DNA fragment containing a fragile site (susceptible to replication difficulties), to study the loss of HR-mediated heterozygocity [54]. Many tumors contain alteration in such sites which lead to cancer progression. In addition, several sporadic and hereditary cancers [55] imply mutations in HR pathways or epigenetic silencing in such genes, yielding variable responses to current chemotherapies and genomic instability [56]. Moreover, tumor cells deficient in repair genes depend on backup systems for their survival. This dependence could be used, from a therapeutic point of view, to induce a synthetic lethality in the tumor. For instance, tumor cells deficient in HR could be treated with DSB inducers; though tumor cells can gain resistance by invoking biochemical mechanisms to reduce drug action or acquiring additional alterations on repair pathways [57].
In summary, the collection of strains constructed for the experiments presented here has a potential use for genotoxicological and anti-genotoxicological evaluation. The dup-seg assay is an ideal candidate for the assessment of compounds that introduces deletions due to unequal exchanges.
ACKNOWLEDGEMENTS
This work was supported by Consejo Nacional de Ciencia y Tecnología [CONACYT 101039]; and Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica, UNAM [PAPIIT IN207513].
CONFLICT OF INTEREST STATEMENT
The authors declare that there are no conflicts of interest.
REFERENCES
1. Blackwood JK, Rzechorzek NJ, Bray SM, Maman JD, Pellegrini L, Robinson NP. End-resection at DNA double-strand breaks in the three domains of life. Biochem Soc Trans. 2013;41(1):314-20.
2. El-Bibany AH, Bodnar AG, Reinardy HC. Comparative DNA damage and repair in echinoderm coelomocytes exposed to genotoxicants. PloS one. 2014;9(9):e107815.
3. Norval M, Lucas RM, Cullen AP, de Gruijl FR, Longstreth J, Takizawa Y, et al. The human health effects of ozone depletion and interactions with climate change. Photochem Photobiol Sci. 2011;10(2):199-225.
4. Yagura T, Makita K, Yamamoto H, Menck CF, Schuch AP. Biological sensors for solar ultraviolet radiation. Sensors. 2011;11(4):4277-94.
5. Sutherland JC. Repair-dependent cell radiation survival and transformation: an integrated theory. Phys Med Biol. 2014;59(17):5073-90.
6. Pascucci B, D’Errico M, Parlanti E, Giovannini S, Dogliotti E. Role of nucleotide excision repair proteins in oxidative DNA damage repair: an updating. Biochemistry (Mosc). 2011;76(1):4-15.
7. Janowska B, Komisarski M, Prorok P, Sokolowska B, Kusmierek J, Janion C, et al. Nucleotide excision repair and recombination are engaged in repair of trans-4-hydroxy-2-nonenal adducts to DNA bases in Escherichia coli. Int J Biol Sci. 2009;5(6):611-20. 8. Cox MM. Recombinational DNA repair of damaged replication forks in Escherichia coli: questions. Annu Rev Genet. 2001;35:53-82.
9. Galitski T, Roth JR. Pathways for homologous recombination between chromosomal direct repeats in Salmonella typhimurium. Genetics. 1997;146(3):751-67.
10. Espinosa-Aguirre J, Barajas-Lemus C, Hernandez-Ojeda S, Govezensky T, Rubio J, Camacho-Carranza R. RecBCD and RecFOR dependent induction of chromosomal deletions by sodium selenite in Salmonella. Mutat Res. 2009;665(1-2):14-9.
11. Luria SE, Delbruck M. Mutations of bacteria from virus sensitivity to virus resistance. Genetics. 1943;28(6):491-511.
12. Aravind L, Makarova KS, Koonin EV. Survey and summary: Holliday junction resolvases and related nucleases: identification of new families, phyletic distribution and evolutionary trajectories. Nucleic Acids Res. 2000;28(18):3417-32.
13. Saintigny Y, Makienko K, Swanson C, Emond MJ, Monnat RJ, Jr. Homologous recombination resolution defect in werner syndrome. Mol Cell Biol. 2002;22(20):6971-8.
14. Baharoglu Z, Bradley AS, Le Masson M, Tsaneva I, Michel B. ruvA Mutants that resolve Holliday junctions but do not reverse replication forks. PLoS Genet. 2008;4(3):e1000012.
15. Giraud-Panis MJ, Lilley DM. Structural recognition and distortion by the DNA junction-resolving enzyme RusA. J Mol Biol. 1998;278(1):117-33.
16. Zechiedrich L, Osheroff N. Topoisomerase IB-DNA interactions: X marks the spot. Structure. 2010;18(6):661-3.
17. Connelly JC, de Leau ES, Leach DR. DNA cleavage and degradation by the SbcCD protein complex from Escherichia coli. Nucleic Acids Res. 1999;27(4):1039-46.
18. Palchevskiy V, Finkel SE. A role for single-stranded exonucleases in the use of DNA as a nutrient. J Bacteriol. 2009;191(11):3712-6.
19. Eykelenboom JK, Blackwood JK, Okely E, Leach DR. SbcCD causes a double-strand break at a DNA palindrome in the Escherichia coli chromosome. Mol Cell. 2008;29(5):644-51.
20. Miesel L, Roth JR. Salmonella recD mutations increase recombination in a short sequence transduction assay. J Bacteriol. 1994;176(13):4092-103.
21. McGlynn P, Lloyd RG. Genome stability and the processing of damaged replication forks by RecG. Trends Genet. 2002;18(8):413-9.
22. Lopez CR, Yang S, Deibler RW, Ray SA, Pennington JM, Digate RJ, et al. A role for topoisomerase III in a recombination pathway alternative to RuvABC. Mol Microbiol. 2005;58(1):80-101.
23. Grove JI, Harris L, Buckman C, Lloyd RG. DNA double strand break repair and crossing over mediated by RuvABC resolvase and RecG translocase. DNA Repair. 2008;7(9):1517-30.
24. Lovett ST, Hurley RL, Sutera VA Jr., Aubuchon RH, Lebedeva MA. Crossing over between regions of limited homology in Escherichia coli. RecA-dependent and RecA-independent pathways. Genetics. 2002;160(3):851-9.
25. Dutra BE, Sutera VA, Jr., Lovett ST. RecA-independent recombination is efficient but limited by exonucleases. Proc Natl Acad Sci USA. 2007;104(1):216-21.
26. Swingle B, Markel E, Costantino N, Bubunenko MG, Cartinhour S, Court DL. Oligonucleotide recombination in Gram-negative bacteria. Mol Microbiol. 2010;75(1):138-48.
27. Poteete AR. Expansion of a chromosomal repeat in Escherichia coli: roles of replication, repair, and recombination functions. BMC Mol Biol. 2009;10:14.
28. Ivankovic S, Dermic D. DNA end resection controls the balance between homologous and illegitimate recombination in Escherichia coli. PLoS One. 2012;7(6):e39030.
29. Cardenas PP, Carrasco B, Defeu Soufo C, Cesar CE, Herr K, Kaufenstein M, et al. RecX facilitates homologous recombination by modulating RecA activities. PLoS Genet. 2012;8(12):e1003126.
30. Sakai A, Cox MM. RecFOR and RecOR as distinct RecA loading pathways. J Biol Chem. 2009;284(5):3264-72.
31. Morimatsu K, Wu Y, Kowalczykowski SC. RecFOR proteins target RecA protein to a DNA gap with either DNA or RNA at the 5’ terminus: implication for repair of stalled replication forks. J Biol Chem. 2012;287(42):35621-30.
32. Lehmann AR, Niimi A, Ogi T, Brown S, Sabbioneda S, Wing JF, et al. Translesion synthesis: Y-family polymerases and the polymerase switch. DNA repair. 2007;6(7):891-9.
33. Shukla AK, Roy KB. Rec A-independent homologous recombination induced by a putative fold-back tetraplex DNA. Biol Chem. 2006;387(3):251-6.
34. Seigneur M, Ehrlich SD, Michel B. RuvABC-dependent double-strand breaks in dnaBts mutants require recA. Mol Microbiol. 2000;38(3):565-74.
35. Al-Hadid Q, Ona K, Courcelle CT, Courcelle J. RecA433 cells are defective in recF-mediated processing of disrupted replication forks but retain recBCD-mediated functions. Mutat Res. 2008;645(1- 2):19-26.
36. Belle JJ, Casey A, Courcelle CT, Courcelle J. Inactivation of the DnaB helicase leads to the collapse and deg¬radation of the replication fork: a com-parison to UV-induced arrest. J Bacteriol. 2007;189(15):5452-62.
37. Khan SR, Kuzminov A. Replication forks stalled at ultraviolet lesions are rescued via RecA and RuvABC protein-catalyzed disintegration in Escherichia coli. J Biol Chem. 2012;287(9):6250-65.
38. Ishioka K, Fukuoh A, Iwasaki H, Nakata A, Shinagawa H. Abortive recombination in Escherichia coli ruv mutants blocks chromosome partitioning. Genes Cells. 1998;3(4):209-20.
39. Jaktaji RP. Using transposon mutagenesis to find an alternative resolvase in an Escherichia coli cells lacking RuvABC. Pakistan J Biol Sci. 2009;12(6):534-7.
40. Jeiranian HA, Courcelle CT, Courcelle J. Inefficient replication reduces RecA-mediated repair of UV-damaged plasmids introduced into competent Escherichia coli. Plasmid. 2012;68(2):113-24.
41. Courcelle J, Carswell-Crumpton C, Hanawalt PC. recF and recR are required for the resumption of replication at DNA replication forks in Escherichia coli. Proc Natl Acad Sci USA. 1997;94(8):3714-9.
42. Sutherland BM, Bennett PV, Sidorkina O, Laval J. Clustered damages and total lesions induced in DNA by ionizing radiation: oxidized bases and strand breaks. Biochemistry. 2000;39(27):8026-31.
43. Bichara M, Meier M, Wagner J, Cor¬donnier A, Lambert IB. Postreplication repair mechanisms in the presence of DNA adducts in Escherichia coli. Mutat Res. 2011;727(3):104-22.
44. Reams AB, Kofoid E, Kugelberg E, Roth JR. Multiple pathways of duplication formation with and without recombination (RecA) in Salmonella enterica. Genetics. 2012;192(2):397-415.
45. Killoran MP, Keck JL. Sit down, relax and unwind: structural insights into RecQ helicase mechanisms. Nucleic Acids Res. 2006;34(15):4098-105.
46. Hua X, Huang L, Tian B, Hua Y. Involvement of recQ in the ultraviolet damage repair pathway in Deinococcus radiodurans. Mutat Res. 2008;641(1-2):48-53.
47. Kovacic L, Paulic N, Leonardi A, Hodnik V, Anderluh G, Podlesek Z, et al. Structural insight into LexA-RecA* interaction. Nucleic Acids Res. 2013;41(21):9901-10.
48. Hishida T, Han YW, Shibata T, Kubota Y, Ishino Y, Iwasaki H, et al. Role of the Escherichia coli RecQ DNA helicase in SOS signaling and genome stabilization at stalled replication forks. Genes Dev. 2004;18(15):1886-97.
49. Nakayama H, Nakayama K, Nakayama R, Irino N, Nakayama Y, Hanawalt PC. Isolation and genetic characterization of a thymineless death-resistant mutant of Escherichia coli K12: identification of a new mutation (recQ1) that blocks the RecF recombination pathway. Mol Gen Genet. 1984;195(3):474-80.
50. Thoms B, Borchers I, Wackernagel W. Effects of single-strand DNases ExoI, RecJ, ExoVII, and SbcCD on homologous recombination of recBCD+ strains of Escherichia coli and roles of SbcB15 and XonA2 ExoI mutant enzymes. J Bacteriol. 2008;190(1):179-92.
51. Feng WY, Hays JB. DNA structures generated during recombination initiated by mismatch repair of UV-irradiated nonreplicating phage DNA in Escherichia coli: requirements for helicase, exonucleases, and RecF and RecBCD functions. Genetics. 1995;140(4):1175-86.
52. Ivancic-Bace I, Vlasic I, Salaj-Smic E, Brcic-Kostic K. Genetic evidence for the requirement of RecA loading activity in SOS induction after UV irradiation in Escherichia coli. J Bacteriol. 2006;188(14):5024-32.
53. Jockovich ME, Myers RS. Nuclease activity is essential for RecBCD recombination in Escherichia coli. Mol Microbiol. 2001;41(4): 949-62.
54. Casper AM, Rosen DM, Rajula KD. Sites of genetic instability in mitosis and cancer. Ann N Y Acad Sci. 2012;1267:24-30.
55. Evers B, Helleday T, Jonkers J. Targeting homologous recombination repair defects in cancer. Trends Pharmacol Sci. 2010;31(8):372-80.
56. Helleday T. Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis. 2010;31(6):955-60.
57. Bouwman P, Jonkers J. The effects of deregulated DNA damage signaling on cancer chemotherapy response and resistance. Nat Rev Cancer. 2012;12(9):587-98.
Received in December 2013.
Accepted in November 2014.
Elizabeth B Cuétara. Department of Clinical Experimental Pharmacology, Research Division, National Institute of Oncology and Radiobiology, INOR Calle 29 y F, Vedado, CP 10 400, La Habana, Cuba. E-mail: ecuetara@infomed.sld.cu.


{kind=link}
{kind=link}