










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMEDISAN
versión On-line ISSN 1029-3019
MEDISAN v.15 n.3 Santiago de Cuba mar. 2011
ARTÍCULO ORIGINAL
Aspectos clinicoepidemiológicos de la leucemia linfoide crónica
Clinical epidemiological aspects of chronic lymphoid leukaemia
MsC. Marisol Rodríguez Brunet, 1 MsC. Geldris P. Hernández Galano, 2 MsC. Lidia C. Suárez Beyríes, 3 MsC. Beatriz O. de la Uz Ruesga 4 y Dr. Ernesto Duverger Magdaleón 5
1Especialista de I Grado en Hematología y Medicina General Integral. Máster en Enfermedades Infecciosas. Profesora Asistente. Hospital General Docente "Dr. Juan Bruno Zayas Alfonso", Santiago de Cuba, Cuba.
2Especialista de I Grado en Hematología. Máster en Enfermedades Infecciosas. Hospital General Docente "Dr. Juan Bruno Zayas Alfonso", Santiago de Cuba, Cuba.
3Especialista de I Grado en Hematología y Medicina General Integral. Profesora Asistente. Máster en Enfermedades Infecciosas. Hospital General Docente "Dr. Juan Bruno Zayas Alfonso", Santiago de Cuba, Cuba.
4Especialista de I Grado en Hematología y Medicina General Integral. Profesora Asistente. Máster en Urgencias Médicas. Hospital General Docente "Dr. Juan Bruno Zayas Alfonso", Santiago de Cuba, Cuba.
5Especialista de I Grado en Hematología y Medicina General Integral. Hospital General Docente "Dr. Juan Bruno Zayas Alfonso", Santiago de Cuba, Cuba.
RESUMEN
Se hizo un estudio descriptivo y retrospectivo de 71 pacientes con leucemia linfoide crónica, atendidos en el Servicio de Hematología del Hospital General Docente "Dr. Juan Bruno Zayas Alfonso" de Santiago de Cuba, desde enero del 2001 hasta noviembre del 2006, con vistas a identificar algunas variables clinicoepidemiológicas en ellos, mostrar las terapéuticas más empleadas, así como evaluar la supervivencia, las principales causas de la entidad clínica y la mortalidad. En la serie predominaron los ancianos, el sexo masculino y la categoría de alto riesgo correspondiente a los estadios avanzados. El esquema terapéutico de clorambucil y prednisona fue el más empleado, con buenos resultados en la mayoría de los integrantes de la casuística. La supervivencia de los pacientes, en general, osciló entre 1-5 años, en tanto las muertes ocurridas fueron causadas por progresión de la enfermedad, procesos infecciosos respiratorios, transformación prolinfocítica, segundas neoplasias y accidentes vasculares encefálicos.
Palabras clave: leucemia linfoide crónica, esquemas terapéuticos, estadificación, supervivencia, mortalidad, Servicio de Hematología.
ABSTRACT
A descriptive and retrospective study of 71 patients with chronic lymphoid leukemia, attended at the Hematology Service from ¨Dr Juan Bruno Zayas Alfonso¨ Teaching General Hospital in Santiago de Cuba was carried out from January, 2001 to November, 2006, in order to identify some clinical epidemiological variables on them, to show the therapeutical variables more used, as well as to assess survival, mortality, and the main causes of the clinical entity. Elderly, male sex, and high risk category related to advanced stage were predominant in the series. The therapeutical schedule of chlorambucil and prednisone was the most used, achieving good results in the majority of the case material. The survival of patients, in general, ranged among 1-5 years, whereas deaths occurred due to disease progression, infectious respiratory processes, pro-lymphocytic transformation, second neoplasias, and strokes.
Key words: chronic lymphoid leukemia, therapeutical schedules, staging, survival, mortality, Hematology Service.
Recibido: 22 de septiembre de 2010
Aprobado: 27 de octubre de 2010
INTRODUCCIÓN
La leucemia linfoide crónica (LLC) es una enfermedad caracterizada por la proliferación y acumulación de linfocitos B neoplásicos, que se hallan mayormente en fase Go del ciclo celular. Las manifestaciones clínicas se deben a la infiltración progresiva de estos linfocitos atípicos (que tienen una vida media más larga) en la médula ósea, los ganglios linfáticos y otros tejidos, así como a las alteraciones inmunológicas que invariablemente se asocian a la citada afección.1,2
De hecho, la incidencia de esta entidad clínica, que es la forma más frecuente de leucemia en los países occidentales -- donde representa alrededor de 30 % --, aumenta con la edad y varía según el área geográfica. Su causa se desconoce; pero a diferencia de lo que ocurre en otras leucemias, no existe relación entre la LLC y la exposición a radiaciones ionizantes, agentes químicos y virus, aunque sí parece haberla, según algunos autores, 1,2 con factores genéticos e inmunológicos y variaciones cromosómicas.
El riesgo de que los familiares de primer grado de una persona con LLC también la padezcan, es 2 a 7 veces mayor que en comparación con otros individuos. Algunos estudios en gemelos univitelinos han mostrado la diversidad genotípica de la leucemia, que la revela como una enfermedad adquirida, fundamentalmente a expensas de las conectivopatías o los estados de inmunodeficiencia (congénita o no). 2 Aproximadamente 50 % de los afectados por leucemia presentan anomalías citogenéticas, con mayor frecuencia la trisomía 12 (numéricas) y las deleciones 11q y del 13q12-14 (estructurales).1
Para diagnosticar la mencionada afección se parte de la forma de los linfocitos en sangre periférica (de aspecto maduro) e inmunofenotipo (CD 19+, CD5+, CD23+, CD10-, bcl-1-), si bien este perfil inmunológico puede compartir algunas características con otros procesos linfoproliferativos. Al respecto, el diagnóstico no indica que necesariamente deba prescribirse determinado tratamiento, pues en algunos pacientes existe poca masa tumoral y su esperanza de vida no se acorta sustancialmente por dicha morbosidad. Tales personas serán tratadas solamente si se encuentran sintomáticas o se comprueba que la enfermedad progresa hacia un estadio más avanzado.2-4
En las 2 últimas décadas se han producido importantes avances en el conocimiento de este tipo de leucemia, tanto desde el punto de vista biológico como de su génesis. La identificación de numerosos factores de pronóstico, así como el resultado de diversos ensayos terapéuticos, permiten formular toda una serie de recomendaciones útiles en la práctica.
Por lo antes expuesto y con el propósito de trazar estrategias sanitarias futuras, basadas en el riesgo individual, no solo para paliar los síntomas y mejorar la calidad de vida de estos pacientes, sino además para convertir en realidad la esperanza de lograr devolver la salud en algunos casos, se propuso realizar la presente investigación.
MÉTODOS
Se hizo un estudio descriptivo y retrospectivo de 71 pacientes con leucemia linfoide crónica, atendidos en el Servicio de Hematología del Hospital General Docente "Dr. Juan Bruno Zayas Alfonso" de Santiago de Cuba, desde enero del 2001 hasta noviembre del 2006, con vistas a identificar algunas variables clinicoepidemiológicas en ellos, señalar las terapéuticas más empleadas, así como evaluar la supervivencia, las principales causas de la entidad clínica y la mortalidad.
La enfermedad se diagnosticó a través de recuentos linfocitarios en sangre periférica (SP), estudio morfológico o anatomopatológico de médula ósea, o ambos (mediante aspiración o biopsia, respectivamente). Se consideraron los criterios de diagnóstico utilizados por el International Workshop on Chronic Lymphocytic Leukemia (IWCLL) 1 y el National Cancer Institute and Sponsored Working Group. 3
De las historias clínicas correspondientes se extrajeron las siguientes variables de interés: edad, sexo, estadio clínico, fecha de diagnóstico, esquema de tratamiento recibido, respuesta clínica, complicaciones y fecha de fallecimiento. Los datos fueron procesados mediante la prueba de proporción y se empleó el porcentaje como medida de resumen.
Se utilizó la clasificación integrada de Binet y Rai, recomendada por el IWCLL, 1 para la estadificación de la leucemia linfoide crónica en cada paciente:
Estadios clínicos de Binet
| a) Estadio A: | Ausencia de anemia y trombocitopenia (Menos de 3 áreas "linfoides fectadas") |
| b) Estadio B: | Ausencia de anemia y trombocitopenia |
| c) Estadio C: | Anemia (Hb<100g/L) o trombocitopenia (o ambas) |
Se consideraron 5 "áreas linfoides": hígado, bazo y ganglios linfáticos de las zonas cervicales, axilares e inguinales, tanto unilaterales como bilaterales.
Estadios clínicos de Rai
| a) Bajo riesgo: | Estadio 0 | Solo linfocitosis >15 000/mm3 en SP, >40 % linfocitos en médula ósea |
| b) Riesgo intermedio: | Estadio I Estadio II |
|
| c) Riesgo alto: | Estadio III Estadio IV | Linfocitosis + anemia (Hb<110g/L) Linfocitosis + trombopenia (<100 000/ mm3) |
El Internacional Workshop on CLL aconseja integrar la clasificación de Binet y Rai como sigue: estadios A (O), A (I), A (II), B (I), B (II), C (III) y C (IV), donde los precoces fueron A, O, I; y los avanzados: B, C, II, III y IV.
Los esquemas de poliquimioterapia incluyeron el tratamiento convencional con clorambucil más prednisona (o sin esta) u otros patrones de quimioterapia intensiva como CHOP (ciclofosfamida, adriamicina, vincristina y prednisona) y COP (ciclofosfamida, vincristina y prednisona), por citar algunos, de modo que las bases terapéuticas quedaron así establecidas:
- Estadio de bajo riesgo (BR): No se administró tratamiento alguno, salvo que se detectaran manifestaciones clínicas de progresión patológica.
- Estadio de riesgo intermedio (RI): Se utilizó quimioterapia intensiva cuando se presentaron manifestaciones clínicas de progresión patológica; pero algunos pacientes no requirieron ser tratados de inmediato.
- Estadio de alto riesgo (AR): Se indicó quimioterapia intensiva.
La terapéutica también se justificó cuando hubo categorías de AR (RAI- II/IV o Binet C), manifestaciones clínicas de progresión de la enfermedad, síntomas B, signos de duplicación linfocitaria durante un período menor de 12 meses y citopenias inmunes refractarias.
De acuerdo con el IWCLL 1 se definieron las siguientes respuestas:
- Remisión completa (RC): Ausencia de síntomas y signos atribuibles a la enfermedad, número de linfocitos en sangre periférica menor de 4x109/L, más de 100 plaquetas x109 /L, así como resultados normales de la aspiración y biopsia medular.
- Remisión parcial (RP): Cambio de la enfermedad a un estadio menos avanzado luego del plan terapéutico.
- Progresión: Evolución de la enfermedad a otro estadio, como consecuencia del tratamiento.
RESULTADOS
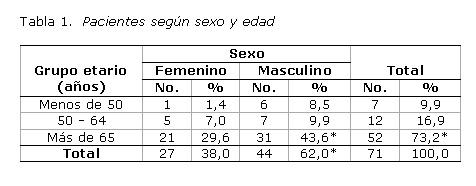
La entidad clínica predominó en los varones (62,0 %), con una relación de 1,5 a 1. La edad de los pacientes al diagnosticar la enfermedad fue de 65 años o más (73,2 %). Solo 9,9 % tenía menos de 50 años (tabla 1).
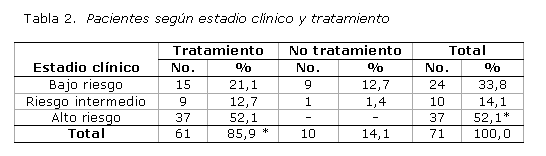
Según el estadio clínico y el tratamiento recibido (tabla 2), predominó la categoría de alto riesgo correspondiente a los estadios avanzados de la enfermedad, con 37 pacientes, para 52,1 %, mientras que los de riesgo bajo e intermedio representaron 33,8 y 14,1 %, respectivamente.
El tratamiento citotóxico fue indicado en 85,9 % de los afectados y solo no estuvo justificado su empleo en 14,1 %. Los pacientes evaluados como alto riesgo (52,1 %) y riesgo intermedio (12,7 %) fueron tratados y exclusivamente 1 afectado de riesgo intermedio (1,4 %) no recibió la terapia por fallecer antes de las 24 horas, como consecuencia de un accidente vascular encefálico. De los pacientes con bajo riesgo, 21,1 % recibió quimioterapia y 12,7 % no la requirió.
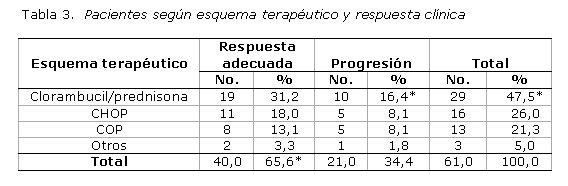
De los esquemas terapéuticos utilizados (tabla 3), la combinación formada por clorambucil/prednisona resultó ser el más empleado (47,5 %) y con mejor respuesta clínica (31,2 %). En cuanto a los pacientes tratados con quimioterapia, 65,6 % mostró una respuesta clínica favorable (completa o parcial), mientras que 34,4 % evolucionó hacia estadios más avanzados de la enfermedad.
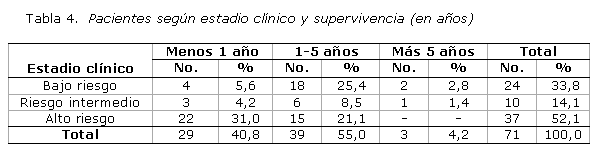
La supervivencia global de los integrantes de la casuística osciló entre 1-5 años, para 55,0 % (tabla 4), mientras que 4,2 % rebasó los 5 años y el resto (40,8 %) no superó este período. Los pacientes en estadios precoces mostraron una mayor supervivencia: entre 1-5 años, con 25,4 %, y más de 5 años, con 2,8 %; los ubicados en estadios avanzados exhibieron cifras inferiores al año (31,1 y 4,2 %, para los de alto riesgo y riesgo intermedio, respectivamente). Ninguno sobrepasó los 5 años.
En la investigación fueron registradas 22 defunciones, para 31 % de mortalidad general. La principal causa de muerte fue la progresión de la enfermedad (54,5 %), seguida de los procesos infecciosos respiratorios (27,3 %). La transformación prolinfocítica, las segundas neoplasias y los accidentes vasculares encefálicos fueron detectados en 1 paciente (4,5 %), respectivamente.
DISCUSIÓN
La leucemia linfoide crónica se presenta comúnmente después de los 50 años de edad, con evidente predominio en el sexo masculino (2:1) e incidencia de 5 personas por cada 100 000 en este grupo etario y 30 por 100 000 habitantes en el grupo de más de 80 años. La edad media de diagnóstico oscila entre 50 y 60 años y se ha señalado 1-5 que solo 10 % de los afectados tiene edades inferiores al ser diagnosticados.
Su incidencia tiene una gran diferencia según el área geográfica, pues varía desde 2,5 % de todas las leucemias en Japón hasta 38 % en Dinamarca; en los países occidentales dicha incidencia anual es de aproximadamente 3 afectados por cada 100 000 habitantes. Por otro lado, constituye la leucemia más frecuente en Europa y Norteamérica (30 % del total y 75 % de las formas crónicas).3
En el pronóstico de la LLC ha significado un gran avance la introducción de los estadios clínicos, pues en cada uno de ellos se incluyen los afectados con un pronóstico heterogéneo, pues este factor está influenciado, a su vez, por otros factores (patrón histopatológico de la médula ósea, cifras de linfocitos en sangre periférica, tiempo de duplicación linfocitario, número de prolinfocitos en sangre periférica, alteraciones citogenéticas). En la actualidad constituyen la guía fundamental para indicar tratamiento.
Este incremento en la supervivencia se debe fundamentalmente al diagnóstico más preciso de la entidad clínica, que separa la enfermedad de otros síndromes linfoproliferativos crónicos, con los cuales, sin duda, era confundida en el pasado; además, gracias a los factores de pronóstico, es posible establecer indicaciones terapéuticas basadas en el riesgo individual de cada paciente, pues mientras que algunos afectados fallecen pocos meses después del diagnóstico, otros sobreviven durante más de 10 años.
Con carácter excepcional, en 1 % de las personas que padecen leucemia linfoide crónica, esta desaparece de forma "espontánea"; en los pacientes con mal pronóstico, la mediana de supervivencia rara vez rebasa los 5-6 años.6-8
El diagnóstico de LLC no implica necesariamente que deba instaurarse tratamiento oncoespecífico. Algunos pacientes poseen poca masa tumoral (estadio A de Binet y 0 de Rai, con infiltración poco intensa de médula ósea, recuentos linfocitarios bajos o estables en sangre periférica) y su esperanza de vida no se acorta sustancialmente por la enfermedad; tales personas no deben ser tratadas a no ser que presenten síntomas o exista progresión a un estadio más avanzado.
Al respecto, el tratamiento precoz en estos pacientes no ha mostrado mejoría de la supervivencia, sin embargo, aumenta el riesgo de desarrollar tumores sólidos. En estudios aleatorios se ha comprobado que tienen una duración de 10-15 años (sin problemas clínicos importantes), la que no es diferente de los grupos control comparables, por tanto, debe realizarse una observación inicial sin terapéutica, pues la enfermedad permanece estable, sin indicios de progresión durante años, en aproximadamente 20-30 % de los pacientes (LLC quiescente o indolente). 1,2
Por el contrario, el pronóstico de los pacientes con estadios iniciales avanzados (B y C de Binet o II, III y IV de Rai) con una mediana de supervivencia de 18-36 meses, obliga a iniciar de manera precoz un tratamiento.4,5
El promedio de supervivencia de pacientes con leucemia linfática crónica de alto riesgo es de 2 años desde el momento del diagnóstico. Los afectados en estadio avanzado debido a una alta masa tumoral con infiltración de la médula ósea, tienen peor pronóstico que aquellos con estadios avanzados de mecanismo inmune; por ello en cada estadio se incluyen pacientes que muestran supervivencia de tan solo unos meses y otros cuya esperanza de vida no se ve modificada por la afección.
De hecho, el pronóstico se deriva del estadio de la enfermedad. Algunos de los afectados tienen igual supervivencia que la de cualquier persona con la misma edad que no padezca LLC; no obstante, en otros estadios más avanzados, y en dependencia de los factores pronósticos, la supervivencia puede ser considerablemente inferior y también estará en relación con las complicaciones que puedan surgir en la evolución de esta leucemia. 9-12
Los resultados de la serie coinciden con lo señalado por otros autores, 13, 14 quienes describen las infecciones como una de las complicaciones más frecuentes debido a la disminución de la inmunidad del organismo por la propia enfermedad, las que también son secundarias a la reducción de las defensas del cuerpo por los tratamientos aplicados. Otras complicaciones lo constituyen los procesos autoinmunitarios (anemia hemolítica), la aparición de otras afecciones tumorales (por ejemplo, melanoma y cáncer de pulmón) y la transformación de la LLC en otro tipo de síndrome linfoproliferativo más "agresivo".
Pueden presentarse diversas dificultades, pues a medida que la enfermedad evoluciona, aumentan las posibilidades de las complicaciones subsiguientes al incremento de la masa tumoral, por lo que resulta frecuente la aparición de trombocitopenia y anemia, consecutivas a la insuficiencia medular. Muchos pacientes adquieren un estado de inmunodeficiencia en algún momento de su evolución, casi siempre atribuible a infecciones bacterianas (74 %), localizadas sobre todo en los pulmones o el tracto genitourinario, o virales (21 %), generalmente producidas por herpesvirus; estas últimas específicamente incrementadas en quienes se utiliza fludarabina.15
Con menor frecuencia se detectan infecciones micóticas, entre ellas la meningitis criptocócica e histoplasmosis diseminada; recientemente se ha planteado la posibilidad de que se instalen micosis sistémicas, sobre todo las producidas por Candida albicans o Aspergillus. Se ha señalado, además, que las infecciones constituyen la principal causa de mortalidad (comprendida entre 30 y 50 %) y de morbilidad en la LLC, pues llegan a producirse aproximadamente en 80 % de los pacientes.16-18
Este estado de inmunodeficiencia predispone a la aparición de segundas neoplasias, por ello cuando un paciente con LLC presente nuevos síntomas o toma del estado general, sin que exista una explicación adecuada, debe pensarse en la posibilidad de una neoplasia asociada que, generalmente, puede ser cutánea, del tubo digestivo o de pulmón. También se ha comunicado la asociación de algunas hemopatías mieloides, entre ellas: leucemia mieloide crónica, policitemia vera, trombocitemia esencial, leucemia mieloblástica y síndromes mielodisplásicos, pero sin que se haya establecido una relación causal entre los procesos. 19,20
REFERENCIAS BIBLIOGRÁFICAS
1. American Cancer Society. Cancer Facts and Figures 2010. Atlanta: American Cancer Society, 2010.
2. Dighiero G, Hamblin TJ. Chronic lymphocytic leukaemia. Lancet 2008; 371(9617):1017-29.
3. Hallek M, Cheson BD, Catovsky D. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 2008; 111(12):5446-56.
4. Shanafelt TD, Kay NE, Jenkins G. B-cell count and survival: differentiating chronic lymphocytic leukemia from monoclonal B-cell lymphocytosis based on clinical outcome. Blood 2009; 113(18):4188-96.
5. Rawstron AC, Bennett FL, O'Connor SJ. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med 2008; 359(6):575-83.
6. Dighiero G. Monoclonal B-cell lymphocytosis-a frequent premalignant condition. N Engl J Med 2008; 359(6):638-40.
7. Shanafelt TD, Kay NE, Rabe KG. Brief report: natural history of individuals with clinically recognized monoclonal B-cell lymphocytosis compared with patients with Rai o chronic lymphocytic leukemia. J Clin Oncol 2009; 27(24):3959-63.
8. Landgren O, Albitar M, Ma W. B-cell clones as early markers for chronic lymphocytic leukemia. N Engl J Med 2009; 360(7):659-67.
9. Moreno C, Villamor N, Colomer D. Allogeneic stem-cell transplantation may overcome the adverse prognosis of unmutated VH gene in patients with chronic lymphocytic leukemia. J Clin Oncol 2005; 23(15):3433-8.
10. Wierda WG, O'Brien S, Wang X. Prognostic nomogram and index for overall survival in previously untreated patients with chronic lymphocytic leukemia. Blood 2007; 109(11):4679-85.
11. Moreton P, Kennedy B, Lucas G. Eradication of minimal residual disease in B-cell chronic lymphocytic leukemia after alemtuzumab therapy is associated with prolonged survival. J Clin Oncol 2005; 23:2971-9.
12. Foon KA, Hallek MJ. Changing paradigms in the treatment of chronic lymphocytic leukemia. Leukemia 2010; 24:500-11.
13. Keating MJ, O'Brien S, Albitar M. Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab as initial therapy for chronic lymphocytic leukemia. J Clin Oncol 2005; 23(18):4079-88.
14. Kantarjian H, O'Brien S. The chronic leukemias. In: Goldman L, Ausiello D. Cecil Textbook of Medicine. 23 ed. Philadelphia: Saunders Elsevier, 2007.
15. Grever M, Andritsos LA, Lozanski G. Chronic lymphoid leukemia. In: Abeloff MD, Armitage JO, Niederhuber JE, Kastan MB, McKenna WG. Abeloff's Clinical Oncology. 4 ed. Philadelphia: Elsevier Churchill Livingstone, 2008.
16. Corte Arboleya Z, Zakariya-Yousef Breval F, Lequerica Fernández P, Ferreiro Artime N, Gutiérrez Cecchini B, Venta Obaya R. Interés clínico de la electroforesis de proteínas. Boletín Informativo del Hospital San Agustín (Asturias) 2008; 9(1).
17. Harris NL, Jaffe ES, Diebold J. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting-Airlie House, Virginia, November 1997. J Clin Oncol 1999; 17(12):3835-49.
18. Rai KR, Döhner H, Keating MJ, Montserrat E. Chronic lymphocytic leukemia: case-based session. Hematology 2001. Education Program. Orlando: American Society of Hematology, 2001:140-56.
19. Kay NE, Hamblin TJ, Jelinek DF, Dewald GW, Byrd JC, Farag S, et al. Chronic lymphocytic leukemia. Hematology 2002. Education Program. Philadelphia: American Society of Hematology, 2002:193-213.
20. Döhner H, Stilgenbauer S, Benner A. Genomic aberrations and survival in chronic lymphocytic leukemia. N Eng J Med 2000; 343:1910-6.
MsC. Marisol Rodríguez Brunet. Hospital General Docente "Dr. Juan Bruno Zayas Alfonso", avenida Cebreco, km 1½, reparto Pastorita, Santiago de Cuba, Cuba.
Dirección electrónica: MsC. Marisol Rodríguez Brunet

{kind=link}
{kind=link}
{kind=link}
{kind=link}