










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Endocrinología
versión On-line ISSN 1561-2953
Rev Cubana Endocrinol v.14 n.2 Ciudad de la Habana Mayo-ago. 2003
Enfoque actual
Hospital Docente "Calixto García"
Obesidad: fisiología, etiopatogenia y fisiopatología
Dra. Lidia Esther Rodríguez Scull1
Resumen
La obesidad se ha convertido en un serio problema de salud a nivel mundial, por su estrecha vinculación con las principales causas de morbimortalidad. La relación obesidad-resistencia insulínica-diabetes mellitus-hipertensión arterial adquiere cada día mayor importancia, por el papel cada vez más relevante de la obesidad en el desarrollo de cada una de ellas. Los conocimientos sobre la fisiología del mantenimiento del peso corporal, a partir de la ruptura de los mecanismos fisiológicos que dan origen a la obesidad, el descubrimiento y la caracterización de la leptina y sus mediadores, así como los factores ambientales perpetuadores de un fenómeno que tiene causas genéticas, pero que a la vez tiene condicionadores ambientales socioculturales, dan origen a este trabajo con el que nos proponemos profundizar en el tema de la obesidad como un fenómeno multifactorial con graves consecuencias clínicas.
Palabras clave: Obesidad; genética; ambiente.
La obesidad es una condición patológica, muy común en el ser humano y presente desde la remota antigüedad, que persiste y se incrementa durante siglos por factores genéticos y ambientales, hasta convertirse actualmente en una pandemia con consecuencias nefastas para la salud.1
Si se evalúa la figura humana en el tiempo a través de la imagen plástica, se comprueba que probablemente la obra más antigua de arte plástica conocida, representa una figura femenina con una obesidad mórbida: la Venus de Willendorf, que no es una sola sino más de cien esculturas, la mayoría de figuras obesas y que evidencian la presencia de la obesidad desde hace más de 25 000 años. Los griegos plasmaron con precisión el cuerpo humano y en sus obras la obesidad no es un hecho frecuente, más bien priman las figuras delgadas. Sin embargo, a medida que las sociedades se extienden y con ellas el arte pictórico, que no es más que su reflejo, la figura humana es cada vez más plena, lo que se muestra en las esculturas y pinturas de Miguel Angel y Rafael, Rubens, Velázquez y Goya por citar algunas. Y es que el hombre, en su largo camino a la "civilización", ha abandonado, adquirido o reformado costumbres que, junto a una base genética favorable, ha originado la explosión de un fenómeno que es cada vez más serio y peligroso por sus consecuencias: la obesidad.
En la actualidad se estima el número de personas obesas en el mundo en más de 300 millones,2 con una amplia distribución mundial y una prevalencia mayor en países desarrollados o en vías de desarrollo.3-7,9,10 Este incremento en la prevalencia de proporciones epidémicas está relacionado con factores dietéticos y con un incremento en el estilo de vida sedentario.11 El aumento del consumo de grasas saturadas y de carbohidratos, la disminución de la ingestión de frutas, vegetales y pescado, así como de la actividad física, que se expresa desde la ausencia de esta actividad programada hasta el incremento del tiempo dedicado a actividades con notable base sedentaria, como ver la televisión o trabajar en la computadora, son las causas más importantes en el desarrollo de este problema de salud mundial.12 Por otra parte, las consecuencias de la obesidad alcanzan proporciones catastróficas. El riesgo de muerte súbita de los obesos es tres veces mayor que el de los no obesos, y es el doble para el desarrollo de insuficiencia cardíaca congestiva (ICC), enfermedad cerebrovascular (ECV) y cardiopatía isquémica (CI), mientras la posibilidad de desarrollar de diabetes mellitus (DM) es 93 veces mayor cuando el índice de masa corporal (IMC) pasa de 35.13 Por otra parte, la obesidad tiene una relación estrecha con la resistencia a la insulina y con factores genéticos y ambientales probablemente comunes. La resistencia a la insulina tiene efectos fisiopatogénicos importantes en el desarrollo de DM, síndrome metabólico, e HTA.14
Las investigaciones realizadas a lo largo de las dos últimas décadas han revolucionado los conocimientos sobre los mecanismos fisiológicos y moleculares que regulan la grasa y el peso corporal. La clonación de los genes que corresponden a los síndromes de obesidad monogenética y la caracterización de las vías determinadas a través de estos puntos de entrada genéticos, el descubrimiento de la leptina, su receptor y el receptor de la melanocortina,4 así como la comprobación, con la utilización de la biología molecular de la acción de diversos mediadores hormonales implicados en el mantenimiento del peso corporal, han contribuido a la comprensión de los elementos fisiológicos de este, así como a los factores etiológicos y patogénicos de la obesidad. A esto debe añadirse el gran salto dado por las ciencias epidemiológicas y farmacológicas con los estudios evidenciales, y la participación de forma decisiva en la comprensión del fenómeno y de sus consecuencias, con la búsqueda de pautas para su solución.
Por la importancia que adquiere la obesidad como fuente de graves problemas, asociada a las principales causas de muerte y discapacidad, al caudal enorme de conocimientos que sobre este aspecto se ha acumulado y el estímulo que puede significar para su estudio, control y prevención, nos proponemos hacer una revisión acerca de los mecanismos que la originan y perpetúan.
Antecedentes
La obesidad se define como un exceso de grasa corporal o tejido adiposo.10 Desde el punto de vista práctico se considera el índice de masa corporal (IMC) el método ideal para el diagnóstico de la obesidad, por su buena correlación con la grasa corporal total.
El IMC es igual al peso corporal en kilogramos, dividido entre la talla en metros cuadrados (IMC = peso en kg/ talla en m2). Se considera ideal un IMC entre 20 y 25; sobrepeso entre 25 y 29,9; obesidad grado I de 30 a 34,9 de IMC; obesidad grado II de 35 a 39,9 de IMC y obesidad grado III, extrema o mórbida, con un IMC mayor de 40.15 Esta clasificación no es arbitraria, sino el resultado de estudios que demuestran que por encima de 25 de IMC aumentan las probabilidades de eventos relacionados con la enfermedad aterosclerótica y sus consecuencias, como son los cardiovasculares y cerebrovasculares, y las alteraciones metabólicas como la resistencia a la insulina, la diabetes mellitus, las alteraciones de los lípidos y la hipertensión arterial, sin mencionar las neoplasias y los trastornos del tractus gastrointestinal.13
Existen otras formas para diagnosticar la obesidad, como la medición de los pliegues cutáneos en diferentes sitios, con ecuaciones y nomogramas para la conversión del grosor del pliegue en grasa y que se expresa en el porcentaje de grasa corporal que debe ser no mayor de 28 % en la mujer y no mayor del 20 % en el hombre. Se requieren cuatro pliegues para estas mediciones, que son los del bíceps, tríceps, subescapular y suprailíaco, aunque es también útil la medición de solo dos. Una forma menos complicada es la utilización aislada del tríceps, que se considera normal en la mujer por debajo de 30 mm y en el hombre de 23 mm. Esto tiene su explicación a partir de la consideración de que aproximadamente el 50 % de la grasa corporal se encuentra en el tejido celular subcutáneo.16,17 La medición de los pliegues tiene el inconveniente de que la distribución de la grasa difiere en individuos con igual cantidad de tejido adiposo y que en ciertas formas de obesidad, la grasa tiene una distribución generalizada, mientras en otras es fundamentalmente abdominal. Por otra parte, la relación grasa subcutánea/grasa profunda (visceral) puede ser de 0,1 a 0,7, además de que la grasa corporal aumenta con la edad, no así el grosor del pliegue.18 También existen otros métodos como son la medición de la densidad corporal por isótopo-dilución, la conductividad eléctrica bajo el agua, la tomografía axial computarizada y la resonancia magnética nuclear, que son directos y precisos, pero complicados, poco prácticos y costosos, confinados por eso a la investigación. Además, no consideran el carácter anatómico, la distribución de la grasa y las consecuencias clínicas, que es lo que brinda valor pronóstico.19
La medida del índice cintura - cadera, al ser expresión de la cantidad de grasa intra-abdominal, ha adquirido un valor predictivo importante de riesgo de alteraciones y consecuencias metabólicas de la obesidad, por lo cual su uso como diagnóstico de obesidad casi iguala en importancia al IMC. Se determina dividiendo la circunferencia a nivel del ombligo y el máximo de circunferencia de las caderas y los glúteos. Este índice es mayor en el hombre que en la mujer, precisamente por la distribución de la grasa en ambos sexos y tiende además a aumentar con la edad. Un índice mayor de 0,95 en el hombre y de 0,80 en la mujer es predictor de aumento del riesgo de anormalidades metabólicas;20,21 Sin embargo, en los últimos años es considerada la circunferencia de la cintura el mejor marcador de sobrepeso y obesidad, por expresar una relación muy estrecha con la grasa abdominal, responsable en mayor medida, de las consecuencias metabólicas directas relacionadas con la obesidad.15,21 Una circunferencia de la cintura mayor de 94 mm en el hombre y de 80 mm en la mujer, es diagnóstico de sobrepeso u obesidad abdominal aun cuando el IMC no lo evidencie, y resulta un marcador importantísimo de futuras complicaciones; por lo tanto, es un punto de partida para la intervención médica, sobre todo si se asocian otros factores de riesgo como la hipertensión arterial (HTA), la diabetes mellitus (DM), o las alteraciones lipídicas (HLP).21-24
Fisiología
De acuerdo con la primera ley de la termodinámica, la obesidad es el resultado del desequilibrio entre el consumo y el aporte de energía.19 La energía que el organismo utiliza proviene de 3 fuentes: carbohidratos, proteínas y grasas. La capacidad de almacenar carbohidratos en forma de glucógeno, igual que la de proteínas, es limitada. Solo los depósitos de grasas se pueden expandir con facilidad para dar cabida a niveles de almacén superiores a las necesidades. Los alimentos que no se consumen como energía, se almacenan, y por lo tanto, es la grasa la principal fuente de almacén y origen de la obesidad. Los carbohidratos son el primer escalón en el suministro de energía. Cuando el consumo de carbohidratos excede los requerimientos, estos se convierten en grasas. En ausencia o con niveles muy bajos de glúcidos, y con necesidades energéticas presentes, las proteínas a través de los aminoácidos son utilizadas para la producción de energía o para la movilización, utilización y almacenamiento de las grasas, proceso conocido como gluconeogénesis, en el cual los aminoácidos con esqueleto de carbono son convertidos, por múltiples reacciones, en piruvato, que a su vez va a derivar en glucosa. Esta glucosa neoformada es oxidada o utilizada para la formación de triglicéridos mediante su conversión a glicerol.25 Las grasas que se ingieren son utilizadas primeramente como fuente de almacén en forma de triglicéridos en el adiposito, o para la producción de hormonas y sus componentes celulares. Una vez que los almacenes primarios de energía hayan agotado sus reservas fácilmente disponibles, son las grasas las encargadas de suministrar la energía necesaria y se movilizan de sus depósitos, proceso en el cual participan activamente las proteínas.26
De este modo, el cuerpo humano cumple las leyes físicas representadas por este primer principio de la termodinámica, según el cual la energía ni se crea ni se destruye, solo se transforma. Todo exceso de energía introducida cambia la energía interna del organismo y se transforma en energía química, y como principal almacén está el tejido graso. Un ingreso energético (IE) mayor que el gasto o consumo energético total (CET), inevitablemente causará un aumento del tejido adiposo, que siempre se acompaña del incremento de la masa magra, así como también del peso corporal, en cuyo control el CET desempeña una función importante.
El CET guarda relación con la masa magra corporal y la mezcla metabólica oxidada está relacionada con los alimentos ingeridos, la capacidad de adaptación del cuerpo y la velocidad de consumo energético. Para mantener el equilibrio energético, es necesario oxidar la mezcla de combustible ingerida. Cualquier desviación ya sea mayor o menor, provocará un desbalance.
Por lo tanto, el peso corporal puede variar en relación con la ingestión (IE) y/o el GET, que es igual al consumo energético en reposo o basal (CEB) más el consumo energético durante la actividad física (CEA) más el consumo energético en la termogénesis (CET):27
CET = CEB + CEA + CET.
El consumo energético basal (CEB) representa hasta el 70 % del CET19 y depende, a su vez, del peso corporal total, del período en que se encuentre el individuo ya sea ayuno, sobrealimentado, en restricción dietética u obeso, porque para cualquiera de estos estados existe un sistema de regulación preciso, cuya función es mantener el peso corporal. Las variaciones en el peso corporal llevan aparejados cambios en el CEB. El aumento de peso se produce en 2/3 a expensas del tejido adiposo, y 1/3 de masa magra; el CEB de estos tejidos es de 5 cal/kg y 40cal/kg, respectivamente27 y como el aumento de peso no es solo dependiente del tejido graso, se produce invariablemente un aumento del gasto energético encaminado al mantenimiento del nuevo equilibrio establecido por el sistema. Pero mientras no existe un límite superior para la ganancia a expensas del tejido graso, sí lo hay para la masa magra, que es de hasta 100 kg en el hombre y 70 en la mujer,19 de forma que, una vez llegado a ese límite, futuras ganancias de peso serán a expensas del tejido adiposo. Lo contrario ocurre con la pérdida de peso, que aunque está basada en la pérdida de grasa, también se pierde masa no grasa, lo que provocará una caída del consumo energético, proporcional a la pérdida de estas, cuyo fin es mantener el equilibrio. Es decir, las variaciones en el consumo energético basal que dependen del peso corporal, imponen un ritmo para mantener este último, pero a su vez determinan, junto al ingreso energético, ganancias o pérdidas; mientras mayor es el peso corporal a expensas de tejido graso por aumento del ingreso energético, menor es el consumo energético, y un consumo energético basal bajo es un buen predictor de futuras ganancias de peso.28 La cantidad de energía consumida durante la actividad física representa el 20 % del GET y está en relación con el peso corporal y con la edad, con la cual esta tiende a disminuir, así como con el IG.19,27 Para un IG estable, los cambios en el nivel de la actividad física traen como consecuencia variaciones en el peso corporal. De este modo, la actividad física representa la forma de gasto más variable de la ecuación, de forma que aunque represente aproximadamente el 20 % del GET, puede llegar a ser el 80 % como se ve en los deportistas de alto rendimiento.27 El efecto termoenergético de los alimentos está constituido por el gasto en la masticación, tránsito, digestión, absorción y metabolismo y por el efecto termogénico de los alimentos en forma de termogénesis adaptativa, ambos controlados por el sistema simpático, y determina el 10 % restante del GET.29 Una forma peculiar de termogénesis es la producida por el hábito de fumar, y es por eso que el abandono de este debe ir acompañado de una disminución del ingreso con vista a evitar una ganacia de peso provocada por una disminución del CET.27 La termogénesis adaptativa es una forma de gasto energético en forma de calor que tiene lugar en el tejido adiposo pardo, y que cumple un importante papel en algunos mamíferos, sobre todo en los que hibernan, y que el hombre en su largo camino evolutivo casi lo perdió y quedó confinado solo a los recién nacidos y a los adultos en una mínima proporción. El tejido adiposo pardo o marrón es altamente especializado en la producción de calor. Está muy vascularizado, y en sus mitocondrias la llamada proteína de desacoplamiento de la grasa parda UCP1 desacopla la fosforilación oxidativa, y el resultado de esto es la conversión de energía en calor.
Recientemente se han descubierto dos nuevas proteínas de desacoplamiento UCP 2 y 3 que se expresan en más alto grado en el humano adulto.30 Este tejido tiene una importante inervación simpática y su papel termogénico se ve incrementado específicamente por la estimulación de los Beta 3 receptores exclusivos del tejido graso, y su estimulación produce cambios en su estructura, lo que promueve la generación de calor en respuesta al frío y la ingesta.27 Por otra parte, la deficiencia en roedores de este tejido, produce obesidad. En el hombre, la distribución y la cantidad es muy escasa y su papel en la obesidad está en estudio.
En la regulación del gasto energético y de la ingesta participan el sistema nervioso, el sistema digestivo y el adiposito. Este último será abordado, en primer lugar y de forma especial, porque un cambio en la concepción de esta célula de solo almacenador de energía en forma de triglicéridos, hacia la comprensión de este como todo un órgano, ha revolucionado los estudios y el manejo de la obesidad como enfermedad.31
El adipocito es una célula altamente diferenciada con tres funciones: almacén, liberación de energía y endocrino metabólica. Puede cambiar su diámetro veinte veces, y su volumen mil. Deriva de su precursor: el adipoblasto, indistinguible a simple vista del fibroblasto, y es identificado por genes y proteínas específicas, como el factor gamma de proliferación y activación capaz de llevar los fibroblastos indiferenciados a diferenciarse como adipositos.19 El adiposito secreta una serie de sustancias con funciones diversas y con implicaciones clínicas importantes, como son: factor de necrosis tumoral alfa, proteína C, molécula de adhesión intercelular, factor de VWV, angiotensinógeno, inhibidores del activador del plasminógeno 1, adiponectin, resistin, etc.32 Es, sin embargo, el descubrimiento de la leptina y de los genes que regulan su producción desde el adiposito, lo que ha originado la gran revolución en el conocimiento de la regulación ingesta-gasto y, por lo tanto, en la evaluación de la obesidad aun cuando el camino por recorrer es todavía largo.
La leptina es la señal aferente de grasa mejor conocida y el mejor candidato a ser la fundamental señal de comunicación al sistema nervioso central de la información sobre la grasa corporal.
Esta citosina producida fundamentalmente por el tejido adiposo, pero también en menor medida por la placenta y el estómago, disminuye la ingestión de alimentos e incrementa el gasto energético. Este péptido ejerce sus efectos a través de un receptor: el de la leptina, ubicado en las neuronas del núcleo infundibular del hipotálamo, con las siguientes consecuencias:
- Disminución de la secreción de neuropéptido Y, que es el más potente estimulador del apetito.
- Disminución de la secreción de la proteína relacionada con el agutí. En inglés Agouti related protein, descrita primeramente en roedores, en los cuales las mutaciones dominantes originan obesidad, resistencia a la insulina, hiperleptinemia y color amarillo, y que fue posteriormente caracterizada en el hipotálamo humano. Esta proteína es un antagonista de los receptores de la melanocortina 1 y 4, que son reguladores del apetito.
- Aumento de la secreción de la propia melanocortina, el precursor de la hormona alfa melanotropina, que reduce la ingestión de alimentos.
- Aumento de la secreción de producto peptídico regulado por cocaína- anfetamina (CART), que produce un incremento del gasto y una disminución de la ingestión.
La leptina, además de estas vías, a través del hipotálamo utiliza el sistema nervioso simpático para sus efectos por su estimulación en la liberación de tirotropina,33 pero el sistema nervioso simpático no participa en la regulación del gasto ni de la ingesta; solo por mediación de la leptina, los receptores noradrenérgicos también modulan el peso corporal. La estimulación de lo receptores alfa1 y beta 3 por la noradrenalina disminuye la ingesta y aumenta el consumo energético, mientras que la acción sobre otros tipos de receptores, como los alfa 2A, 2B y 2C, tienen un efecto contrario.34
El sistema nervioso parasimpático eferente (vagal), por su parte, modula el metabolismo hepático, la secreción de insulina y el vaciamiento gástrico, y participa también en el control del peso corporal. La disminución de la glucemia precede hasta el 50 % de las comidas en los animales y de los seres humanos. Cuando este fenómeno, que es independiente del nivel de partida del descenso de la glucosa, se bloquea, se retrasa la toma de alimentos.35
Los estímulos olfatorios y gustativos producidos por el alimento participan en la regulación de la ingesta. Todas estas señales periféricas son integradas en el sistema nervioso con la consecuente liberación de neurotransmisores. Estos neurotransmisores pueden aumentar o disminuir la ingestión de alimentos, y muchos tienen especificidad para macronutientes. De ellos uno de los más estudiados es la serotonina. Los receptores de la serotonina modulan tanto la cantidad de alimento como la selección de los macronutrientes. La estimulación de estos en el hipotálamo reduce la ingestión en general y de las grasas en particular, con poco efecto sobre carbohidratos y proteínas.36 El neuropéptido Y aumenta la ingestión de alimentos y es el más potente de los neutransmisores en la acción anabólica.33,37 El sistema de la melanocortina y los receptores opiáceos también reducen la ingestión con especificidad para las grasas.38
Por su parte, los péptidos intestinales modulan también la cantidad de alimentos. Por ejemplo, la colecistocinina, el péptido liberador de gastrina, la neuromedina b y la bombesina39-41 disminuyen la ingestión de alimentos. El péptido afín al glucagón, producido por las células L del intestino, es un muy potente insulinótropo, al estimular la secreción de insulina por las células beta del páncreas dependiente de la ingesta, así como su neogénesis y la biosíntesis de proinsulina. Tiene además la capacidad de disminuir la secreción de glucagón, el vaciamiento y la secreción gástrica, lo que con disminución de la concentración de glucosa en sangre y de la respuesta a la insulina lleva a un incremento de la sensación de saciedad y una disminución de la ingesta.6,29,42,43 El páncreas endocrino ofrece la insulina como hormona reguladora del peso y del metabolismo por excelencia, lo que favorece la utilización de la glucosa y los lípidos por los tejidos, disminuye la producción hepática de glucosa, y como resultado de esto proporciona la optimización en el empleo de las proteínas al balancear positivamente el anabolismo.
El glucagón, también producido por el páncreas, estimula la degradación del glucógeno y la gluconeogénesis44 lo que favorece el catabolismo. Por su parte, la porción exocrina aporta la enterostatina (señal peptídica de la colipasa pancreática) la cual disminuye la ingestión de grasa y produce saciedad.45
Los sistemas eferentes de control del peso corporal son el motor para la adquisición de alimentos, el endocrino y el neurovegetativo.
El sistema endocrino está representado por las hormonas del crecimiento, las tiroideas, las gonadales, los glucorticoides y la insulina.
Durante la etapa del desarrollo, la hormona del crecimiento y las tiroideas trabajan al unísono para aumentar el crecimiento. En la pubertad comienzan a funcionar los esteroides gonadales, los que provocan desplazamiento en la proporción de la grasa respecto al peso corporal magro en niños y niñas. La testosterona aumenta el peso corporal magro y en relación con la grasa y los estrógenos tienen un efecto contrario. Los niveles de testosterona disminuyen cuando el varón humano se hace mayor, y provocan un aumento de la grasa visceral y corporal total, con disminución del peso corporal magro. Con la edad, esto se complica con la disminución de la hormona del crecimiento, que se acompaña de aumento de la grasa corporal.46
Los glucorticoides suprarrenales tienen una acción importante en el control neuroendocrino de la toma de alimentos y el consumo energético, y son cruciales para el desarrollo y el mantenimiento de la obesidad.47
La insulina es un importante modulador del peso corporal por su acción lipogénica y antilipolítica, y por su papel en el desarrollo de la obesidad.48
El sistema neurovegetativo completa el círculo en el control del peso como regulador de las secreciones hormonales y de la termogénesis.46 Cuando todos estos sistemas, señales y genes funcionan correctamente y están bien modulados por un ambiente favorable, el peso corporal permanece estable o con pocas variaciones anuales. Cuando este equilibrio de fuerzas se quiebra por motivos diversos, aparece la obesidad (fig. 1).
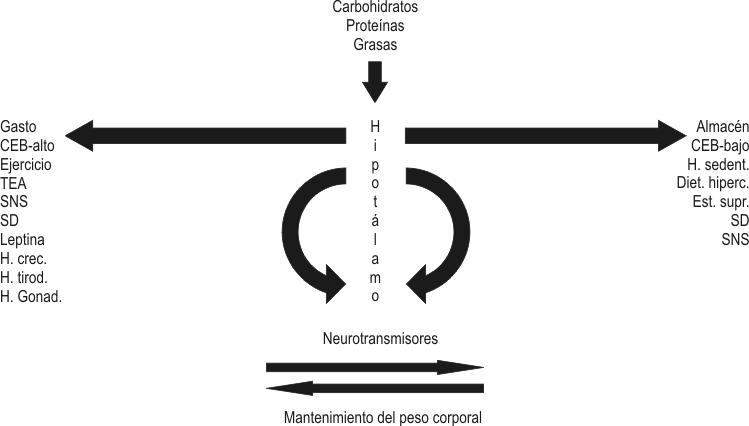
CEB: Gasto energético basal.
TEA: Termogénesis adaptativa.
SNS: Sistema nervioso simpático.
SD: Sistema digestivo.
H .CREC: Hormona del crecimiento.
H. TIROD: Hormona tiroidea.
H. GONAD: Hormonas gonadales.
H. SEDENT: Hábito sedentario.
EST. SUPR: Esteroides suprarrenales.
DIET HIPERC: Dieta hipercalórica.
FIG. 1. Mantenimiento del peso corporal y equilibrio de factores.
Etiopatogenia de la obesidad
La proporción y cantidad de alimentos ingeridos, como carbohidratos, proteínas y grasas, está destinada a convertirse en energía y en elementos celulares, o a almacenarse en forma de grasa.
Los elementos de la columna izquierda de la figura 1 favorecen el gasto energético y el mantenimiento o la pérdida de peso corporal. los de la derecha, disminuyen el gasto, promueven el almacén de energía y con esto la obesidad. Ambos están influenciados por los neurotransmisores, y en ellos el sistema nervioso simpático tiene activa participación.
Con los conocimientos actuales de la fisiología, la genética, la biología molecular y los estudios epidemiológicos evidenciales, podemos establecer que la etiopatogenia de la obesidad es un fenómeno complejo. A simple vista, la teoría de un aumento crónico de la ingesta en relación con el gasto es simple, ya que la obesidad es un trastorno especifico y heterogéneo por su origen, en el cual están implicados factores genéticos y ambientales.
Factores genéticos
La identificación de la mutación ob en ratones genéticamente obesos ob/ob, representa el punto de partida documentado de la acción de los genes en la obesidad. Estos ratones desarrollan obesidad, insulino-resistencia, hiperfagia y un metabolismo eficiente (engordan con la misma dieta que los ratones delgados). El gen ob es el responsable de la producción de leptina y se expresa igualmente en humanos, lo que es descrito en varias familias con obesidad temprana, acompañada de alteraciones neuroendocrinas como hipogonadismo hipogonadotrópico. Lo mismo sucede con la mutación del gen db responsable de la codificación del receptor de la leptina y también encontrada en humanos.49
Existen otras evidencias de la participación de los genes en el origen de la obesidad como son: mutaciones en el gen humano que codifica la proopiomelanocortin (POMC), produce obesidad severa por fallo en la síntesis de alfa MSH, el neuropéptido que se produce en el hipotálamo, e inhibe el apetito. La ausencia de POMC causa insuficiencia suprarrenal por déficit de la hormona Adrenocorticotrópica (ACTH), palidez cutánea y pelo rojo por ausencia de alfa MSH.50 Otros estudios genéticos en roedores muestran varios candidatos para mediadores moleculares de la obesidad. El gen fat codifica la carboxipeptidasa E, una enzima procesadora de péptidos, que participa en el procesamiento de hormonas y neuripéptidos, y la mutación de este gen causa obesidad en ratones.51 La proteína relacionada con el agutí (AGRP) se expresa con el NPY en el hipotálamo y antagoniza la acción de la alfa MSH en los receptores MC4; la mutación del gen agutí produce obesidad por una expresión ectópica de la proteína relacionada con el agutí.52 Por otra parte, una mutación en los genes que codifican el peroxisome - proliferator activated receptor gamma (PPAR gamma) un factor de trascripción del adiposito necesario para la adipogénesis, ha sido relacionado con la obesidad en individuos alemanes.53 Dos síndromes raros, pero conocidos y con base genética, tienen entre sus componentes fundamentales la obesidad: el síndrome de Prader Willi, que se caracteriza por baja estatura, retraso mental, hipogonadismo hipogonadotrópico, hipotonía, pies y manos pequeñas, boca de pescado e hiperfagia, y en la mayoría de los casos tiene una delección del cromosoma 15 y el síndrome de Laurence-Moon Biedl, con retraso mental, retinosis pigmentaria, polidactilia e hipogonadismo hipogonadotrópico.54 Lo mismo sucede con otros síndromes raros con base genética como son los de Alstron, de Bardet-Biedl, de Carpenter y de Cohen.
Todos estos hechos, junto a la evidencia de que los gemelos homocigóticos, aun cuando crezcan separados, sus pesos siempre son parecidos y que el peso de los hijos casi siempre es parecido al de sus padres biológicos, incluso cuando hayan sido adoptados, apoyan el papel de los genes en la etiología de la obesidad. A su vez, los familiares de primer grado de los individuos con obesidad de comienzo en la niñez, tienen el doble de probabilidades de ser obesos que aquellos con obesidad de comienzo en la adultez. Además, aun cuando la obesidad más frecuente no siga un patrón mendeliano, parece ser que los genes contribuyen hasta en un 30 % en el nivel de grasa visceral, no así a la subcutánea.55 También está el hecho de que una predisposición genética a la obesidad pudiera ser el resultado de la herencia de una eficiencia metabólica alta, ya que el nivel de metabolismo basal tiene un componente genético.56 Después de ajustar la tasa metabólica para tejido magro, edad y sexo, el 40 % de variación restante también tiene un importante componente genético.57 En resumen, todo parece indicar que en la mayoría de los casos, la obesidad responde a la interacción de múltiples genes y del ambiente.
Factores ambientales
La evidencia de que el hambre evita o revierte la obesidad, aun en las personas con gran carga genética, junto a su incremento en los países industrializados o en vías de desarrollo en los cuales la dieta es rica en grasas y carbohidratos, y los hábitos sedentarios han aumentado con el desarrollo económico, se muestra a favor del factor ambiental en su origen y desarrollo.58 Otro hecho relevante lo representa el incremento epidémico de la obesidad en los últimos veinte años, que no puede ser explicado por alteraciones genéticas poblacionales desarrolladas en tan corto tiempo. Por otra parte, a medida que la pobreza ha disminuido en países industrializados o en vías de desarrollo, ha aumentado en forma proporcional la obesidad. Lo cierto es que el desarrollo económico trae aparejado problemas sociológicos importantes: aumenta la vida sedentaria al disponerse de medios de transporte, elevadores, equipos electrodomésticos diversos, incluido el uso del control remoto y el tiempo destinado a ver televisión o trabajar en el computador; a su vez, la actividad física programada, aunque se practique quizás con mayor frecuencia e intensidad, no es siempre por los más sedentarios, y la mayoría de las veces está seguida de un período de actividad sedentaria. También se suman la comercialización de comidas altas en calorías y bajas en nutrientes, el aumento en la frecuencia de estas por su fácil accesibilidad, el aumento en la ingestión de grasas saturadas y la disminución en el aporte de comidas sanas, como los vegetales, frutas, pescado y cereales, cuyo costo y tiempo de preparación puede ser mayor que el de la comida fácil. Todo esto, unido a efectos psicológicos de la vida moderna con sus grandes conflictos, ayudan y perpetúan el incremento del fenómeno.
Fisiopatología de la obesidad
Sea cual sea la etiología de la obesidad, el camino para su desarrollo es el mismo, un aumento de la ingestión y/o una disminución del gasto energético.59 Los lípidos procedentes de la dieta o sintetizados a partir de un exceso de carbohidratos de la dieta, son transportados al tejido adiposo como quilomicrones o lipoproteínas de muy baja densidad (VLDL). Los triglicéridos de estas partículas son hidrolizados por la lipoproteinlipasa localizada en los capilares endoteliales, introducidos en el adiposito y reesterificados como triglicéridos tisulares. Durante los períodos de balance positivo de energía, los ácidos grasos son almacenados en la célula en forma de triglicéridos; por eso, cuando la ingestión supera el gasto, se produce la obesidad.60 En la medida en que se acumulan lípidos en el adiposito, este se hipertrofia y en el momento en que la célula ha alcanzado su tamaño máximo, se forman nuevos adipositos a partir de los preadipocitos o células adiposas precursoras, y se establece la hiperplasia. El paciente muy obeso que desarrolla hiperplasia y comienza a adelgazar, disminuirá el tamaño de los adipositos, pero no su número.26 Este hecho tiene una relevancia especial en la obesidad de temprano comienzo, en la niñez o la adolescencia, en la cual prima la hiperplasia sobre la hipertrofia, y como resultado es más difícil su control, pues hay una tendencia a recuperar el peso perdido con gran facilidad y de ahí la importancia de la vigilancia estrecha en el peso de los niños y adolescentes, porque las consecuencias pueden ser graves.
En el caso de la obesidad de comienzo en la adultez, predomina la hipertrofia sobre la hiperplasia, por lo cual su tratamiento suele ser más agradecido, pero no por eso fácil. Por otra parte, se sabe que la distribución de los adipositos y su capacidad de diferenciación, está condicionada genéticamente,31 por eso, mientras mayor sea la fuerza genética para la obesidad, mayor será la probabilidad de que este proceso se desarrolle con el menor esfuerzo y la mayor rapidez.
Tomando en cuenta la leyes de la termoenergética, el paciente obeso debe comer más para mantener su peso, porque además de que su gasto energético es mayor porque el tejido magro también se incrementa con la obesidad, la actividad adrenérgica está estimulada por vía de la leptina, y este aspecto parece ser importante en el mantenimiento de la obesidad.61 Y es que la mayoría de los obesos tienen en realidad una hiperleptinemia con resistencia a la acción de la leptina de forma selectiva, es decir, solo en su capacidad para disminuir la ingestión, pero no en su acción con mediación simpática,62 y por eso el obeso está expuesto no solo a un incremento del gasto mediado por el sistema neurovegetativo, sino también a efectos neuroendocrinos amplificados, con devastadoras consecuencias clínicas. Por eso, cuando se pierde peso a partir de un estado de sobrepeso y/o obesidad, el GEB disminuye, tanto por la misma ley de la termoenergética, como por la disminución de la actividad simpática. De ahí que la pérdida de solo unos pocos kilogramos de peso represente un beneficio multiplicado, por las positivas consecuencias clínicas que esto condiciona, y que las acciones contra la obesidad sean siempre de inestimable utilidad. Los obesos con hipoleptinemia, aleptinémicos o con alteraciones en la acción de los receptores de la leptina, que son el grupo menos numeroso, tienen, por su parte, un gasto energético disminuido con desregulación de los mecanismos controladores de la ingestión que da origen y perpetúa la obesidad, y se ha demostrado que se corrige con la administración de leptina recombinante en el caso de las alteraciones de la leptina, no así en los problemas del receptor.
Otro hecho importante lo constituye el envejecimiento en su amplio sentido de ganancia en años vividos, ya que cuando ocurre este se pierde masa magra, que si no es balanceado con una disminución de la ingesta, lleva a la ganancia de peso lenta e irremediablemente. Además, aunque el aumento de peso está mediado por ambos tejidos (magro y graso), hay que recordar que, llegado el límite superior de crecimiento del tejido magro, todo aumento posterior depende de la grasa cuyo gasto energético es menor, por lo cual el GET tiende a estabilizarse o disminuir de acuerdo con el punto inicial, y si el IE permanece igual, habrá más ganancia de peso (fig. 2).

FIG. 2. El incremento de la ingestión de carbohidratos y grasas, unido a la disminución del gasto energético, fundamentalmente por aumento del sedentarismo, aunque también por alteraciones genéticas en relación con la leptina, originan la obesidad y sus consecuencias. La hiperleptinemia con resistencia es importante en el desarrollo de estas.
Summary
Obesity has turned into a serious health problem worldwide because of its close link to the main causes of mortality and morbidity. The obesity-insulin resistance-diabetes mellitus-blood hypertension relationship has a greater importance today due to the ever increasingly relevant role of obesity in the onset of each of these entities. Knowledge about the physiology of body weight maintenance, on the basis of rupture of physiological mechanisms giving rise to obesity, the discovery and characterization of leptin and its mediators as well as the environmental factors that perpetuate a phenomenon which has genetic causes and are at the same time sociocultural environmental conditioners constitute the main elements of this paper aimed at delving into the topic of obesity as a multi-factor phenomenon with serious clinical consequences.
Key words: OBESITY; GENETICS; ENVIRONMENT.
Referencias bibliográficas
1. Bierman EL. Obesidad. En: Cecil Tratado de medicina interna. 15 ed. La Habana: Pueblo y Educación;1984 p 2030-9.
2. Rossner S. Obesity, the disease of 21th century. Int J Obes 2002;26(suppl 4): S2-S4.
3. Montero JC. Epidemiología de la obesidad en siete países de América Latina. Nutric obes. 2002;5(6):325-30.
4. Booth ML, Chey T, Wake M, Northon K, Hesketh K, Dallman J et al. Change in the prevalence of overweight and obesity among young- australians, 1969-1997. Am J Clin Nutr 2003;77(1):29-36.
5. Katzmarzyk PT. The Canadian obesity epidemic,1985-1998. CMAJ 2002;166(8).
6. Barthel B, Cariou C, Lebos-Soison E, Mamos I. Prevalence of obesity in children: study in the primary Parisian schools. Sante Publ 2001,13(1)7-15.
7. Savra SC, Kauridis Y, Tornaritis M, Epiphaniou-Savra M, Chadjigeargeou C, Kafatos A. Obesity in children and adolescents in Cyprus, prevalence and predisposing factors.Int J Obes Relat Metab Desord 2002;26(8):1036-45.
8. Chinn S,Rona RJ. Prevalence and trends in overweight and obesity in three cross sectional studies of british children. 1974-94. BMJ 2001;322(7274):24-26.
9. Panamerican Health Organization (PAHO). Health in the Americas. N 569. Washington DC:Panamerican Health Organization; Scientific Publication.
10. O'Brien P O, Dixon J. The extent of the problem of obesity. Am J of Surg 2002;184(6):s4-s8.
11. Racette SB, Dusinger S, Deusinger RH. Obesity: Overview of prevalence, etiology and treatment. Phys Ther 2003;83:276-88.
12. Contreras J . La obesidad: una perspectiva sociocultural. Nutric y obes 2002;11(8):997-1001.
13. Calle EE, Thun MJ, Petrelli JM, Rodríguez C, Heath CW Jr. Body- mass index and mortality in a prospective cohort of US adult. N England J Med 2000;342:287-9.
14. Rubio M A. ¿Debemos incluir la determinación de resistin en la práctica clínica? Endocrinología 2003;50(3):91-3.
15. National Health, Lung and Blood Institute Clinical Guidelines on the identifications , evaluations and treatment of overweight and obesity in adult. The evidence report. Obes Res 1998;6 (suppl 2) S 51- S 290.
16. Monterrey Gutiérrez P, Porroto Maury C. Procedimiento gráfico para la evaluación del estado nutricional de los adultos según el IMC. Rev Cubana Aliment Nutr 2001;15(1):62-70.
17. Lohman TG. Skinfolds and body density and their relation to body fitness: a review. Hum Biol 1981;53:181-225.
18. Bray GA. Obesity: basic consideration and clinical approaches. Dis Mon1989;35:449-53.
19. Flier JS, Foster DW. Eating Disorders: Obesity, anorexia nervosa and bulimia. En: William's textbook of Endocrinology. 9 ed. Philadelphia: Sunders company;1998:1061-83.
20. Bray GA. Overweight its risking fate: definition, classification, prevalence and risk. Ann NY Acad Sci 1987;499:14-28.
21. Berdasco Gómez A. Evaluación del estado nutricional del adulto mediante la antropometría. Rev Cubana Aliment Nutr 2002;16(2):146-52.
22. Scarcella C, Depress J P. Treatment of obesity: the need to target attention on high-risk patients characterized by abdominal obesity. Cad Saude Publica 2003;19(suppl 1): S7-S19.
23. Booth M L, Hunter C, Gore C J, Bauman A, Owen N. The relationship between body mass index and waist circumference: implications for estimates of the populations prevalence of overweight. Int J Obes Relat Metab Disord 2000;24(8) 1058-61.
24. Siani A, Cappuccio FP, Barba G, Trevison M, Farinara E, Lacone R, Manzini M, et al. The relationship of waist circumference to blood pressure. The Olivetti Heart Study. Am J Hypertens 2002;15(9):780-6.
25. Marks J, Howard A. La Dieta de Cambridge. Cambridge. Cambridge Export Ltd;. 1997.
26. Laycock JF, Wise P. Disorders of lipid metabolism and obesity. En: Laycock JF. Essential Endocrinology.3 ed. New York: Oxford University Press Inc;1996:338-52.
27. Scopinaro N. The physiology of weight change. Obesity on line, 1997 (fecha de acceso 1999) URL http://www.obesiry-online.com/.
28. Ravussin E, Lilliaja S, Knowler W, et al. Reduced rate of energy expediture as a risk factor for body weight gain. N Engl Med 1998;318:467-72.
29. Jonge L, Bray GA. The thermic effect of food and obesity: a critical review. Obes Res 1997;5:622-31.
30. Recquier D, Bouillaud F. The uncoupling protein homologues: UCP1, UCP2, UCP3, ST UCPT and ST UCP. Biocheem J . 2000;345(PT 2):161-79.
31. Cinti S. Adiposity differentiation and transdifferntiation: plasticity of the adipose organ. J Endocrinol Invest 2002 NOV; 25(10):823-35.
32. Guerrero-Millo M. Adipose tissue hormones. J Endocrinol 2002 NOV, 25(10):855-61.
33. Bjorback C, Hollenberg AN. Leptin and melanocortin signaling in the hypothalamus. Vitam Horm 2002;65:281-311.
34. Leibawits SF. Reciprocal hunger-regulating circuits involving alpha-and beta- adrenergic receptors located respectively in the ventromedial and lateral hypothalamus. Proc Natl Acad Sci USA. 1970;67:1063-70.
35. William E, Clutter, Philip E. Cryer. Hipoglucemia. En: Jay H Stein. Internal Medicine. 4 Ed. St Louis, Missouri: Mosby -Year Book; 1994:p.1424-30.
36. Smith BR, York DA, Bray GA. Hypothalamic infusion of serotonin or serotonin receptor agonist suppressed fat intake in a macronutrients diet, paradigm. Am J Phisiol. 1998; 277:R802-R11.
37. Schawartz MW, Wonds SC, Parte D Jr, Selly RJ, Basking DG. Central nervous system control of food intake. Nature 2000;404:661-7.
38. Qu D, Ludwing DS, Gammeltojt S, et al. A role of melanin-concentrating hormone in the central regulation of feeding behavior. Nature 1996, 21(280):243-7.
39. Gibbs J, Smith GP. Peptides of digestive system and Brain. Model of the cholecystokinin. Ann Endocrinol (París) 1998;49:113-20.
40. Lieverse RJ, Jansen JB, van de Zwan A, et al. Bombesin reduces food intake en lean man by cholecystokinin independent mechanism. J Clin Endocrinol Metab 1993,76:1495- 8.
41. Gutzwiller JP, Drewe J, Hildebrand P, Rossi L, Lauper JZ, Belinger C. Effects of intravenous gastring-releasing peptide on food intake in humans. Gastroenterology 1994;106:1168-73.
42. Flint A, Raben A, Astrup A, Holst JJ. Glucagon-like peptide promotes satiety and suppress energy intake in humans. J Clin Inves 1998,101:515-50.
43. Meier JJ, Gallwitz B, Schmidt WE, Nauck MA. Glucagon-like peptide 1 as a regulator of food intake and body weigth: therapeutic perspective. European journal of Pharmacology. Vol 440. Issue 2-3;2002: p 269-79.
44. Seymour MS. Metabolismo Hepático. En: J H Stein. Internal Medicine. 4 Ed. St Louis Missouri :Mosby Year Book;1994.p.26-7.
45. Erlanson-Albertsson C, York D. Enterostatin, a peptide regulating fat intake. Obes Res. 1997;5:360-72.
46. Vermein Barnes H. Disorders of adolescents growth and development. En: J H Stein . 4 ed . St Louis Missouri: Mosby Year Book;1994: p. 1283-92.
47. Woo R, Kush-Daniels R, Horton ES. Regulations of energy balance. Ann Rev Nutr 1985; 5: 411.
48. Pfeiffer EF. Obesity, islet function and diabetes mellitus. Horm Metab Res. 1974; Suppl 4:143.
49. Zhang Y. Positional cloning of the mouse obese gene and its human's homologue. Nature 1994, 372:425-32.
50. Krude et al. Severe early onset of obesity, adrenal insufficiency and red heir pigmentation caused by POMC mutations in humans. Nat Genet 1998;19(2):155.
51. Chey WY. Regulation of pancreatic endocrine secretion. Int J Pancreatol 1991;9:7-20.
52. Argyroupoulos G, Rankinen T, Neufeld DR, Ricet T, Province MA, Leon AS, et al. A polymorphism in the human agouti related protein is associated with the late onset obesity. J Clin Endocrinol Metab 2002 Sept;87(9):4198-202.
53. Ristow M, Muller-Wieland D, Pfeiffer A, et al. Obesity associated with a mutation in a genetic regulator of adiposity differentiation. N Engl J Med 1998;339:953-9.
54. Nicholls RD et al. Imprinting in Prader Willi and Angelman syndroms. Trend Genet. 1998;14:194.
55. Malleck MJ. Health hazard of obesity and weight control in children, a review of the literature. Am J Public Health. 1983; 73:78-82.
56. Bogurdus C, Lilliaya S, Raviessin E. Genetic effect in resting metabolic rate. N Engl. J Med. 1983;315:96-9.
57. Bouchard C, Trembling A, Nadeau T, et al. Genetic effect in resting and exercise metabolic rate. Metabolism 1989;38:364-8.
58. Barceló Acosta M, Borroto Díaz G. Estilo de vida factor culminante en la aparición y tratamiento de la obesidad. Rev Cubana Invest Biomed 2001;20(4):287-95.
59. Raviessin E, Lilliaja S, Knowler WC, et al. Reduced rate of energy expenditure as a risk factor for body- weight. New Engl J Med 1998;318:467-72.
60. Langhans W. Role of the liver in the metabolic control of eating: what we know and we do not know. Neurosci-Biobehav Rev. 1996;20:145-53.
61. Bray GA. Reciprocal relation of food intake and sympathetic activity: experimental observation and clinical implications. Int J Obes Relat Metab Disord 2000;25(supp l2):S8-S17.
62. Correia ML, Haynes WG, Rahmoun K, Morgan DA, Svitz WI, Mark AL. The concept of selective leptin resistance: evidence from agouti yellow mice. Diabetes 2002 Feb;51(2):439-42.
Recibido: 15 de enero de 2003. Aprobado: 17 de febrero de 2003.
Dra. Lidia Esther Rodríguez Scull. Hospital Docente "Calixto García". Ciudad de La Habana, Cuba.

