










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Ciencias Médicas de Pinar del Río
versión On-line ISSN 1561-3194
Rev Ciencias Médicas vol.18 no.6 Pinar del Río nov.-dic. 2014
ARTÍCULO ORIGINAL
Caracterización clínico genética del síndrome Prader Willi
Clinic genetic characterization of Prader-Willi syndrome
Anitery Travieso Tellez1, Reinaldo Menéndez García2, Deysi Licourt Otero3
1Especialista de Primer Grado en Medicina General Integral y Genética Clínica. Instructora. Centro Provincial de Genética Médica Pinar del Río. Correo electrónico: any0511@princesa.pri.sld.cu
2Especialista de Primer y Segundo Grado en Genética Clínica. Máster en Educación Médica. Profesor Auxiliar. Hospital Pediátrico Provincial Pepe Portilla. Pinar del Río. Correo electrónico: generey@princesa.pri.sld.cu
3Especialista de Primer y Segundo Grado en Genética Clínica. Máster en Atención Integral al Niño. Asistente e Investigador Agregado. Centro Provincial de Genética Médica Pinar del Río. Correo electrónico: eysili@princesa.pri.sld.cu
RESUMEN
Introducción: el síndrome Prader Willi es un desorden genético causado por la pérdida de genes contenidos en la región 15q11-q13 del cromosoma paterno.
Objetivo: describir las características clínicas y genéticas de los pacientes con síndrome Prader Willi.
Material y método: se realizó un estudio descriptivo, de corte transversal, con el universo de 15 pacientes con sospecha de síndrome Prader Willi remitidos a consulta provincial de Genética Clínica durante el año 2013. Se consideraron como variables clínicas los criterios diagnósticos según Holms, y como variables genéticas los resultados de los estudios cromosómicos y moleculares.
Resultados: predominó el sexo femenino en un 66.7%. Las edades estuvieron entre los tres y los 41 años. Los criterios mayores más frecuentes resultaron la obesidad troncular y el retraso del neurodesarrollo en el 100% de los pacientes. Los criterios menores más identificados fueron los disturbios del sueño y las dificultades del lenguaje con un 66.7% cada uno. En ninguno de los casos se detectaron anomalías cromosómicas por cariotipificación. Tres pacientes (60%) presentaron la deleción a nivel de la región 15q11-q13 identificada por la técnica de hibridación in sito con fluorescencia.
Conclusiones: la definición del diagnóstico en la provincia resulta demorada. Se requiere de reevaluación según los criterios clínicos en las diferentes etapas de la vida para diagnóstico de certeza. La presencia de hipotonía neonatal y dificultades en la alimentación son elementos asociados al diagnóstico por deleción 15q11-q13.
DeCS: Síndrome de Prader-Silli/etiología/diagnóstico/genética, Sistema nervioso/crecimiento & desarrollo.
ABSTRACT
Introduction: Prader-Willi syndrome is a genetic disorder caused by deleted or unexpressed genes contained in 15q11-q13 region of paternal chromosome. Objective: to describe clinical and genetic characteristics of patients with Prader-Willi syndrome.
Material and method: a descriptive, cross-sectional study was conducted in 15 patients with Prader-Willi suspect, who were referred to the provincial office of Clinical Genetics during 2013. As clinical variables the diagnostic criteria of Holms were considered and as genetic variables, the results of chromosomal and molecular studies.
Results: female sex prevailed in 66.7%. Ages were between 3 and 41. Most frequent major criteria were troncular obesity and neurodevelopment retardation in 100% of the patients. The minor criteria more identified were: sleep disturbances and speech difficulties (66.7% each one). None of the cases presented chromosomal anomalies because of karyotype classification. Three patients (60%) presented deletion at 15q11-q13 region level, which was identified by hybridization in situ with fluorescence.
Conclusions: in the province a definitive diagnosis was delayed. A reassessment is required according to the clinical criteria during the different stages of life in order to achieve a definitive diagnosis. The presence of neonatal hypotonia and difficulties in feeding are associated elements to the diagnosis of 15q11-q13 deletion.
DeCS: Prader-Willi syndrome/etiology/diagnosos/genetics, Nervous system/growth & development.
INTRODUCCIÓN
El síndrome Prader Willi (SPW) es una enfermedad genética rara, caracterizada por las anomalías del hipotálamo-hipofisarias, que cursa con hipotonía grave durante el período neonatal y los dos primeros años de vida, y con hiperfagia con alto riesgo de desarrollar obesidad mórbida en la infancia y la edad adulta; así como dificultades de aprendizaje y graves problemas de conducta y/o psiquiátricos.1, 2
El trastorno afecta a 1 de cada 25.000 recién nacidos, de ambos sexos y de todas las razas. La mayoría de los casos son esporádicos y la recurrencia familiar es poco frecuente.1 Presentan rasgos faciales característicos (frente estrecha, ojos almendrados, labio superior delgado y boca girada hacia abajo), así como manos y pies muy pequeños. La obesidad es un factor importante que influye en la morbilidad y mortalidad de estos pacientes. Las principales complicaciones son el desarrollo de diabetes mellitus tipo II, hipertensión arterial, insuficiencias respiratorias y trastornos articulares.1-4 El grado de disfunción cognitiva varía ampliamente de un niño a otro. Se asocia con problemas de aprendizaje, del habla y de desarrollo del lenguaje que se agravan aún más por los problemas psicológicos y de comportamiento.1
Su etiología se relaciona con anomalías en una región cromosómica crítica, situada en la zona proximal del brazo largo del cromosoma 15 específicamente 15q11-q13; de tal forma que los genes ubicados en esta zona no se expresan cuando se trata del cromosoma 15 de origen paterno.1
Los expertos coinciden en señalar que el diagnóstico debe basarse en criterios clínicos (Criterios de Holms, 1993, revisados en 2001) confirmados posteriormente mediante análisis genético.5
El diagnóstico precoz, la atención temprana y el manejo interdisciplinario junto con la administración de la hormona del crecimiento han mejorado mucho la calidad de vida de los niños afectados.1, 3-4
Cuba no muestra hasta el momento referencias sobre la incidencia al nacimiento ni la prevalencia de este síndrome. En la literatura solo se encuentran reportes aislados de casos.5-7
En la provincia existe un número de pacientes con diagnóstico probable de síndrome Prader Willi, solo evaluados por el servicio de Genética Clínica sin el adecuado seguimiento por parte de la atención primaria de salud ni de otras especialidades.
Ante la insuficiente disponibilidad de estudios que caractericen la población pinareña afectada con este trastorno se realiza la presente investigación con el objetivo de describir las características clínicas y genéticas del síndrome Prader Willi en Pinar del Río.
Los resultados pretenden aportar al conocimiento de este trastorno poco frecuente por parte de los profesionales pinareños del sector salud, en vistas de manejos médicos cada vez más individualizados que mejoren la calidad de vida de los afectados.
MATERIAL Y MÉTODO
Se realizó un estudio observacional, descriptivo, de corte transversal. Fueron estudiados en consulta de Genética Clínica la totalidad de pacientes (15) con diagnóstico probable de síndrome Prader Willi en Pinar del Río. El único criterio de inclusión para el estudio fue tener impresión diagnóstica de SPW en algún momento de la vida. Por este motivo, la muestra quedó conformada por la totalidad del universo.
Cada paciente y su familia fueron sometidos a exhaustivo interrogatorio y examen físico tomando como referencia los criterios clínicos de Holms. Se estudió con radiografías de columna vertebral y huesos largos. Se exploró con examen de refracción y se cariotipificó con estudio de alta resolución cada caso. Luego de concluida esta fase de la evaluación clínica aquellos pacientes con más alto puntaje según criterios clínicos se sometieron a estudio por técnica de hibridación in sito con fluorescencia (FISH). Se tuvo en cuenta el consentimiento informado de padres y tutores legales. Durante la investigación no hubo conflicto de intereses.
RESULTADOS
En el universo estudiado predominó el sexo femenino para un 66.7%. Las edades estuvieron comprendidas entre los tres y los 41 años con una mediana de 15 años. Los criterios mayores presentes con más frecuencia resultaron la obesidad troncular luego de un aumento brusco y el compromiso global del neurodesarrollo que se identificaron en el 100% de los pacientes, seguidos de la hiperfagia que se presentó en el 86.7%. (Tabla 1)
Por su parte, los criterios menores mayormente identificados fueron los disturbios del sueño y las dificultades en la articulación del lenguaje con un 66.7% cada uno y la baja estatura con relación a la talla familiar en el 60%. Del total de pacientes estudiados, el 46.7% tuvo miopía, esotropia y el 26.7% presentó diagnóstico de alguna variante de escoliosis. (Tabla 2)
Solo cinco casos para un 33.3% reunieron el puntaje requerido para plantear diagnóstico según los criterios clínicos aprobados internacionalmente. (Ver figura)
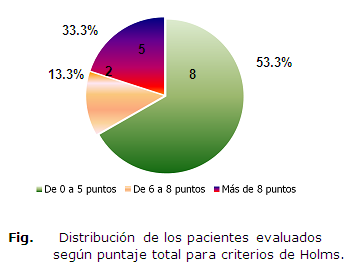
Se muestra que ninguno de los casos presentó anomalía cromosómica en los estudios de alta resolución. De los cinco pacientes con más de siete puntos de acuerdo a los criterios clínicos de Holms, tres (60%) presentaron la deleción a nivel de la región 15q11-q13 identificada por técnica FISH. En este grupo de pacientes los criterios más sensibles fueron la hipotonía neonatal y la pobre succión con dificultades en la alimentación durante la lactancia, elementos que estuvieron presentes en la totalidad de los individuos con la deleción. (Tabla 3)
Finalmente del total de pacientes evaluados solo quedaron con diagnóstico confirmado de SPW cinco pacientes que representa el 33.3% del universo. De ellos dos, solo tienen diagnóstico clínico (13.3%) según puntaje y el resto tienen diagnóstico clínico y molecular (20%) por deleción de la región15q11-q13. La totalidad de este grupo supera los 12 años de edad.
DISCUSIÓN
Sobre el síndrome Prader Willi todos los autores coinciden que su presentación no muestra predominio sobre ningún sexo en particular.1-6 Sin embargo, resulta llamativo que en el presente estudio la mayor parte de los casos se corresponda con el sexo femenino, hallazgo este que no responde epidemiológicamente a ninguna causa particular.
Por su parte, la edad de los pacientes indica la necesidad de evaluar los casos bajo sospecha de diagnóstico de SPW con periodicidad, con el apoyo de los criterios clínicos establecidos.5
Asumir un diagnóstico probable durante la primera infancia podría implicar (como se demostró en este estudio) el riesgo de "etiquetar incorrectamente" un individuo, con la consiguiente desatención de la causa real de sus manifestaciones clínicas. Así mismo, se pierde el sentido de "seguimiento clínico" y no se implementan acciones de prevención terciaria a fin de minimizar las posibles complicaciones del diagnóstico adecuado.
En los pacientes con SPW aparecen con la edad las anomalías por disfunción endocrina: insuficiencia adrenal, hipogonadismo, hipotiroidismo y consecuencias de la obesidad como la diabetes mellitus tipo 2.8 Detenerse en la evaluación de los criterios clínicos aportados por Holms es aceptar la necesidad de seguir los pacientes en el tiempo pues la mayor parte de los criterios no se completan hasta al menos los seis u ocho años de vida.9 No es casual entonces que los pacientes con diagnóstico confirmado en la investigación superen los 12 años de edad.
En 1993, los autores propusieron el consenso de los criterios clínicos para el diagnóstico del SPW. Ocho años más tarde este autor y su equipo de investigadores sugirieron una revisión de estos criterios con vistas a identificar apropiadamente los pacientes tributarios de estudios moleculares confirmatorios.
La propuesta consistió en definir aquellos elementos cardinales según la edad del paciente. De esta forma se estableció la hipotonía neonatal y los problemas de alimentación como principales características de la etapa desde el nacimiento hasta el primer año de vida. Posteriormente y hasta los tres años a estos criterios se incorpora el retardo del neurodesarrollo y el aumento brusco de peso. En la fase de tres y hasta los seis años se añade el hambre insaciable y la obesidad troncular, para finalmente agregar la discapacidad intelectual.10
Reevaluar los casos aplicándoles nuevamente los criterios diagnósticos resultó vital para establecer el diagnóstico clínico de certeza o descartar totalmente el mismo, por lo que se comprueba la variabilidad clínica del trastorno según las diferentes etapas de la vida.9-10
En el presente estudio resulta llamativo que la hipotonía neonatal y la historia de pobre succión con dificultades en la alimentación coinciden en la totalidad de los pacientes con confirmación molecular de la deleción 15q11-q13. Estos resultados apuntan hacia una correlación genotipo-fenotipo particular que coincide con lo expuesto por otros autores9-10 y permiten sugerir que ante un lactante con estos elementos estaría indicado el estudio molecular para diagnóstico de SPW sin necesidad de esperar a otras etapas de la vida, lo que adelantaría la precocidad del diagnóstico.
Finalmente, aquellos pacientes con diagnóstico clínico de certeza y sin presencia de la deleción 15q11-q13, deben corresponder con el resto de las causas genéticas del SPW, entre las que se citan fenómenos de disomía uniparental materna y alteraciones de la impronta genómica.11
REFERENCIAS BIBLIOGRÁFICAS
1. ORPHANET. Sobre las Enfermedades Raras[Internet]. Paris: ORPHANET; Available from: http://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=ES&data_id=139&Disease_Disease_Search_diseaseGroup=S-ndrome-Prader-Willi&Disease [citado 5 Sep 2013]
2. USA. Medlineplus. Síndrome de Prader Willi[Internet]. USA: Medlineplus; Disponible en : http://www.nlm.nih.gov/medlineplus/spanish/praderwillisyndrome.html [citado 5 de septiembre de 2013]
3. Yang L, Zhan G, Ding J, Wang H, Ma D, Huang G, Zhou W. Psychiatric Illness and Intellectual Disability in the Prader-Willi Syndrome with Different Molecular Defects-A Meta Analysis. PLoSOne. [Internet] 2013; 8(8) Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3743792/
4. USA. Johns Hopkins University. PraderWilli Syndrome [Internet]. USA: Johns Hopkins University; 2013. Available from: http://omim.org/entry/176270 [citado 5 Sep 2013]
5. Taboada Lugo N, Lardoeyt Ferrer R. Criterios para el diagnóstico clínico de algunos síndromes genéticos. Rev Cubana Pediatr [Internet]. 2003[citado 24 de enero de 2014]; 75(1). Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034-75312003000100007&lng=es&nrm=iso
6. González Fernández P, Pin Arboledas G, Cabrera Panizo R. Síndrome de Prader-Labhart-Willi y apnea durante el sueño: A propósito de tres pacientes. Rev Cubana Endocrinol [Internet]. 2004[citado 24 de enero de 2014]; 15(2): Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1561-29532004000200006&lng=es
7. Rodríguez Vargas N, Martínez Pérez T, Martínez García R, Calvo Luaces V, Guerrero Guerrero L. Prader-Willi syndrome: Clinical report of two patients and literatura review. Rev Cubana Pediatr[Internet]. 2006[citado 24 de enero de 2014]; 78(1): Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034-75312006000100011&lng=es
8. Emerick JE, Vogt KS. Endocrine manifestations and management of Prader-Willisyndrome. Int J Pediatr Endocrinol[Internet]. 2013[citado 12 enero de 2014]; 1(14). Disponible en http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3751775/
9. Driscoll DJ, Miller JL, Schwartz S, et al. Prader-Willi Syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews[Internet]. Seattle (WA): University of Washington; 1993-2014. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1330/
10. Grechi E, Cammarata B, Chiumello G. Prader-WilliSyndrome: Clinical Aspects. J Obes[Internet]. 2012[citado 24 de enero de 2014]; 2012. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3486015/pdf/JOBES2012-473941.pdf
11. Falaleeva M, Sulsona CR, Stamm S. Molecular Characterization of a patient presumed to have Prader-Willi Syndrome. Clin Med Insights Case Rep[Internet]. 2013[citado nov 2013]; 6: 79-86. Available from: http://www.la-press.com/redirect_file.php?fileId=4941&filename=3659-CCRep-Molecular-Characterization-of-a-Patient-Presumed-to-Have-Prader-Willi-.pdf&fileType=pdf
Recibido: 25 de junio de 2014.
Aprobado: 17 de septiembre de 2014.
Dra. Anitery Travieso Tellez. Especialista de Primer Grado en Medicina General Integral y Genética Clínica. Instructora. Centro Provincial de Genética Médica Pinar del Río. Correo electrónico: any0511@princesa.pri.sld.cu

{kind=link}
{kind=link}
{kind=link}
{kind=link}