










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La hemofilia es una enfermedad hereditaria que impide la correcta coagulación de la sangre y constituye del 80 a 85 % de todas las diátesis hemorrágicas hereditarias. Es producida por la disminución de la actividad del factor VIII (hemofilia A) o del factor IX (hemofilia B). Desde el punto de vista genético se hereda con carácter recesivo ligado al cromosoma X; al final del brazo largo se encuentra el gen del factor VIII (F VIII) en la región Xq28 y el gen del factor IX también se ubica en el brazo largo, pero más cercano al centrómero, en la región Xq27.1
Su incidencia es de aproximadamente 1:5-10 000 varones en el caso de hemofilia A y de 1:30-60 000 en la hemofilia B. Mediante la medición de la actividad funcional de los factores VIII y IX se ha podido clasificar esta enfermedad en severa, menos de 1 % de actividad funcional del factor, presentan hemorragias espontáneas en las articulaciones o músculos y se observa con más frecuencia los sangrados del sistema nervioso central; moderada, del 1 al 5 %, con hemorragias espontáneas ocasionales y suelen ser prolongadas ante traumas o cirugías menores; y leves, entre 6 y 40 % de actividad del factor con presencia de hemorragias graves ante traumatismos o cirugías importantes y las espontáneas son poco frecuentes.2,3) En alrededor de un 25 % de estos enfermos no se encuentran antecedentes familiares, probablemente relacionado con la presencia de nuevas mutaciones.
Desde el punto de vista clínico tanto la hemofilia A como la B son semejantes, en dependencia de la actividad del factor en el plasma será la severidad de las manifestaciones hemorrágicas. Esos eventos recidivantes en los pacientes hemofílicos conllevan una gran morbilidad e influyen notablemente en la calidad de vida de los afectados. El sangrado articular es una característica de esta entidad y la más invalidante. Las hemartrosis repetidas originan una entidad nosológica característica denominada artropatía hemofílica, que provoca limitación severa de la función articular y dolor crónico.4)
Su curso clínico se asocia con gran frecuencia a discapacidades serias del paciente, lo que impacta negativamente en su desarrollo biopsicosocial, así como en la adecuada funcionabilidad familiar. Para muchos niños, padecer esta enfermedad es sinónimo de renunciar a una vida normal de juegos de desarrollo físico, social y hasta profesional. El papel de la familia y el cuidado constante son determinantes en la vida de estas personas.5
El sello distintivo de la hemofilia A son los sangrados recurrentes músculo-articulares, las articulaciones más afectadas son los tobillos, rodillas y codos. También se consideran otras hemorragias que, aunque menos frecuentes, suelen ser muy graves y en ocasiones fatales, como las hemorragias del sistema nervioso central y gastrointestinal.1,2,3) Esos eventos recidivantes, particularmente los musculo-articulares, influyen notablemente en el desenvolvimiento social de los afectados.
El curso de esta entidad clínica se asocia con gran frecuencia a discapacidades graves del paciente que impactan negativamente en su desarrollo, tanto biológico como psicosocial, así como en la adecuada funcionabilidad familiar. Para muchos niños, padecer esta enfermedad es sinónimo de renunciar a una vida normal de juegos, de desarrollo físico, social y hasta profesional. El papel de la familia y el cuidado constante son determinantes en la calidad de vida de estas personas.6
Una complicación relacionada con la terapéutica es la adquisición de inhibidores. El inhibidor es una inmunoglobulina que neutraliza el factor de la coagulación hacia el que está dirigido.7 Son anticuerpos desarrollados por el sistema inmune que se unen al factor administrado y bloquean la eficacia de la terapia de reemplazo lo que dificulta e imposibilita el control de las hemorragias,8) lo que hace ineficaz la estrategia terapéutica y expone a los enfermos a complicaciones importantes asociado a un aumento en los costos del manejo y la morbimortalidad.9 Su presencia es más frecuente en individuos con hemofilia A, con una prevalencia del 12 a 35 %. Se presenta en el 25 al 50 % de los pacientes con hemofilia A severa y usualmente se manifiesta en los primeros 50 días después de comenzado el tratamiento.10
Su severidad, los antecedentes étnicos del individuo, la historia familiar de inhibidores, el genotipo del antígeno leucocitario humano y la mutación que causa la enfermedad, están asociados con el desarrollo de inhibidores; también se describen factores ambientales y la edad al momento de la primera exposición al reemplazo de factor que podría desempeñar algún papel.11,12
Las prioridades a tener en cuenta para mejorar la salud y el bienestar de los pacientes incluyen la prevención de hemorragias y el daño articular. Cuando se produce el sangrado, debe ser tratado lo más precozmente posible con la administración por vía endovenosa del concentrado que contiene el factor plasmático deficiente. La demora en la administración del mismo determina una mayor dificultad en la resolución del cuadro y por tanto, mayor riesgo de secuelas.
Es importante que los pacientes asistan a los talleres educativos que se desarrollan como parte del componente educativo de la terapia de la hemofilia, a fin de incorporar o afianzar conceptos necesarios para un manejo más adecuado del tratamiento. Los centros de atención integral son la mejor opción para las personas con este padecimiento.
Dentro de las modalidades terapéuticas se encuentra la denominada “a demanda”, que consiste en la administración de los concentrados de factor solo ante la aparición de un evento hemorrágico y la “profiláctica”, que es la administración regular de agentes hemostáticos para disminuir o evitar el sangrado de forma permanente, que le permite llevar una vida activa, con una calidad comparable a la de personas sin hemofilia. El tratamiento domiciliario es el ideal para el manejo de los episodios leves (y algunos moderados), para lo cual es importante aprender a reconocer los síntomas iniciales de las hemorragias, determinar su severidad, contar con la medicación en el domicilio, tener capacidad para infundir con la técnica correcta y realizar un uso responsable de los concentrados.13
Al tratarse de una enfermedad ocasio nada por el déficit o la disfunción de una sola proteína, la hemofilia es, en teoría, un candidato ideal para el desa rrollo de estrategias terapéuticas con terapia génica; sin embargo, el F VIII es una glicoproteína de gran tama ño, difícil de expresar en niveles terapéuticos efectivos, condición que interfiere con el desarrollo de este tipo de alternativa.14
En la actualidad, el desarrollo farmacéutico está enfoca do en la generación de productos de acción prolongada que permitan disminuir el número de infusiones administradas al paciente hemofílico, lo que favorece la ad herencia terapéutica del mismo. La ingeniería genética es fundamental en la creación de productos mejorados, como es el caso de la glicosilación, que consiste en fusionar proteínas de manera dirigida, con lo que se ha logrado obtener mejoría del perfil farmacodinámico y prolongar la vida media de proteínas terapéuticas.15
Uno de los mayores desafíos en el tratamiento de la he mofilia es el desarrollo de anticuerpos neutralizantes o inhibidores.10 Estos son aloanticuerpos tipo inmuno globulina G contra los factores de coagulación VIII y IX, que pueden ser inducidos por la terapia sustitutiva con estos factores, los cuales neutralizan rápidamente la acción de los mismos, lo que hace ineficaz la terapia de reemplazo y expone a los pacientes a complicaciones importantes con aumento en los costos del tratamiento.
En este trabajo se describe un caso clínico de un paciente con diagnóstico de hemofilia A moderada y un comportamiento clínico severo, sin inhibidores, que fue tratado con emicizumab, un anticuerpo monoclonal humanizado que se utiliza para restablecer el proceso de la coagulación sanguínea en enfermos con hemofilia, se reportaron resultados satisfactorios en cuanto a la seguridad y la prevención de sangrados.
PRESENTACION DEL CASO
Un adolescente de 18 años de edad, de la piel blanca, atendido en el servicio de Hematología del Hospital Pediátrico Docente Pepe Portilla de Pinar del Río, que, en la etapa neonatal, presentó un hematoma a nivel de muslo derecho después de la administración de la vacuna contra hepatitis B asociado a sangrados por los sitios de puntura.
Los exámenes complementarios realizados mostraron un hemograma con Hb 140 g/L; Hto: 0.43 L/L; plaquetas 305 x 109/L; eritrosedimentación de 10 mm/h y reticulocitos 37 x 10-3. En la lámina periférica se encontró normocromía, normocitosis y plaquetas adecuadas en número, tamaño, forma y agregación. El coagulograma evidenció un TP - control 13 seg / paciente 13 seg; TPT - control 30 seg / paciente 58 seg. Se demostró entonces una disminución de la actividad del F VIII en 1.8 %, lo que demostró el diagnóstico de hemofilia A moderada.
A los tres meses de edad presentó su primera hemartrosis en la rodilla derecha, un evento inusual en esta etapa de la vida por la escasa movilidad. Al año de vida apareció espontáneamente un hematoma que se extendía en toda la cara lateral externa del brazo y antebrazo del miembro superior derecho y a los cinco años presentó hemorragia intracraneal como consecuencia del esfuerzo al vomitar por lo que necesitó cuidados intensivos con uso de altas dosis de concentrado de F VIII.
Por el comportamiento clínico severo de la enfermedad requirió de múltiples ingresos a causa de sangrados articulares, que afectaron principalmente la región del codo derecho, tobillo izquierdo y rodilla derecha, además de hematomas musculares, que le imposibilitaban la deambulación y como consecuencia desarrolló atrofia muscular en ambos miembros inferiores, todo esto lo condujo a un estado de encamamiento durante la mayor parte del día, por lo que la interacción con el medio social, principalmente la escuela, se vio severamente afectada, de ahí que recibiera la actividad docente en el hogar. (Fig. 1)
A los 11 años se inició tratamiento con fisioterapia con la modalidad de hidroterapia con resultados alentadores en la recuperación del tono muscular. A inicios del año 2015, con 12 años de edad, se comenzó a utilizar el régimen de profilaxis con F VIII tres veces a la semana, que, unido a las sesiones de rehabilitación, posibilitaron la disminución de los sangrados, así como la deambulación, se incorporó de forma presencial y sin el apoyo de sillas de ruedas al escenario docente.
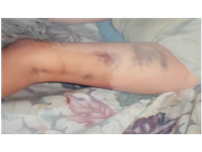
Después de varios años con profilaxis de F VIII y sin el desarrollo de inhibidores, el paciente presentó, aunque con menor frecuencia, hemartrosis principalmente de la rodilla derecha, asociado a dificultad con los accesos venosos para la administración del tratamiento sustitutivo profiláctico, y se decidió, luego de obtenido el consentimiento informado, incorporarlo al régimen de profilaxis con anticuerpo monoclonal emicizumab por vía subcutánea desde julio de 2020. (Fig. 2)
Se inició con una dosis de carga a 3 mg/Kg una vez a la semana durante las cuatro primeras semanas seguido de mantenimiento a 1,5mg/kg semanal, 3mg/kg cada dos semanas o 6mg/kg cada cuatro semanas administrados en forma de inyección subcutánea.
Un mes después de iniciado este tratamiento, el paciente mostró una mejoría sostenida en la tendencia al sangrado articular, sin hemorragias aun después de recibir traumas, así como una buena tolerancia al medicamento sin experimentar reacciones adversas. La tolerancia local fue también satisfactoria, sin equimosis ni zonas eritematosas.

DISCUSIÓN
En los pacientes con hemofilia A, la terapia de profilaxis tiene un alto costo (especialmente con los agentes bypass), cuyo resultado es una corrección fluctuante y parcial de la deficiencia de F VIII con picos en su actividad, por lo que no es completamente eficaz para prevenir todas las hemorragias, ya sean espontáneas o provocadas, además del riesgo de aparición de inhibidores proporcional a la frecuencia de infusión semanal. Otro aspecto a tener en cuenta son las dificultades con los accesos venosos por el número de punciones a los que son sometidos, por lo que es cada vez más difícil el abordaje venoso periférico con el paso de los años.
En este caso, la opción de emplear emicizumab fue una solución acertada, pues disminuyó drásticamente los episodios hemorrágicos, los cuales no habían desaparecido totalmente mientras se mantuvo con profilaxis de F VIII.
Emicizumab es un anticuerpo monoclonal humanizado bioespecífico que se une al factor IX activado y al factor X y sustituye al F VIII en la formación el complejo tenaza, así como su función hemostática, reestablece el proceso de la coagulación sanguínea en los pacientes con hemofilia A.16,17 Es una solución para inyección subcutánea, presentada en bulbos de 0,4; 0,7 o 1 mL que contiene 60, 105 o 150 mg del fármaco.18 Emicizumab fue aprobado inicialmente por la Agencia Europea de Medicamentos y la Food and Drug Administration para su utilización tanto en pacientes adultos como niños, en la profilaxis de los episodios de sangrado en pacientes con hemofilia A grave (deficiencia congénita menor del 1 %) y con inhibidores frente al F VIII.
Actualmente también se ha aprobado para la hemofilia A grave sin inhibidores. Es un agente eficaz para prevenir los sangrados, pero no está indicado para tratarlos.19
Durante el periodo de seguimiento el paciente en cuestión solo presentó un episodio hemorrágico leve, que consistió en un pequeño hematoma en la zona lateral de la rodilla izquierda que se solucionó con un tratamiento de corta duración con F VIII. Tampoco ha presentado efectos adversos destacables, solamente aumento de volumen en la parte inferior de la lengua el primer día de su administración con la dosis de carga, que remitió espontáneamente en una hora. Su cómoda administración por la vía subcutánea hace que este medicamento sea ideal, pues queda abolida la punción venosa como sitio de infusión. Su farmacocinética estable, baja inmunogenicidad y efectiva prevención del sangrado le dan un lugar muy importante para el presente y futuro del tratamiento de las personas con hemofilia A, tanto con inhibidores o sin ellos, con un gran impacto en la vida de estos pacientes.
Los beneficios del mismo son significativos, porque logra activar la coagulación de forma adecuada y permite la formación de un coágulo estable de fibrina, lo cual posibilita que el paciente deje de sangrar. Esta es una nueva modalidad de tratamiento que logró eliminar por completo la tendencia hemorrágica del paciente, principal síntoma de esta dolencia, aspecto que no se había alcanzado anteriormente. Actualmente lleva una vida similar a aquellos niños sin la enfermedad. (Figura 3) Se ha incorporado a la escuela y practica ejercicios físicos, lo cual es muy significativo pues contribuyó a elevar la calidad de vida de este paciente.
Se concluye que el uso del emicizumab en un paciente con hemofilia A sin inhibidores, presentó un elevado perfil de seguridad, así como una alta eficiencia en la prevención de la tendencia hemorrágica. Aunque durante el primer año de tratamiento se produjo un sangrado ligero, aparentemente espontáneo, el mismo no desencadenó ninguna complicación y su resolución fue rápida con la administración de F VIII. La vía subcutánea y su dosificación son una opción atractiva en la profilaxis, pues mejora la reinserción social y la condición clínica de los pacientes con hemofilia A.

