










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El tumor de células gigantes (TCG) recibe esta denominación debido a que contienen gran cantidad de células gigantes de tipo osteoclastos multinucleados, de ahí el nombre de osteoclastoma.
El TCG fue descrito por Cooper en 1818, y redefinido en 1940 por Jaffé y otros,1 conceptuándolo como un tumor localmente agresivo con un componente bifásico de células gigantes y otro fusocelular muy vascularizado, con recidivas frecuentes y una capacidad limitada de producir metástasis.1,2
Es una neoplasia ósea poco predecible en cuanto a su evolución, ya que prácticamente todos los tumores óseos contienen células gigantes,1,3 por lo que se presume que estos tumores pertenecen al estirpe de los monocitos-macrófagos, y se supone que las células gigantes se forman por fusión de las células mononucleares.1,2,3 El objetivo de este trabajo es presentar el caso de un paciente con TCG el cual no es común en nuestro medio y señalar las características diagnósticas de este tumor al mismo tiempo que se revisan los métodos imagenológicos recientes para su confirmación.
Caso clínico
Paciente de 19 años de edad, negro, estudiante universitario, sin antecedentes patológicos familiares ni personales de interés. Llegó a consulta por dolor en la porción distal de la rodilla y el muslo izquierdos desde hacía 2 años e inflamación de dicha zona que le impedía incluso caminar adecuadamente. Acudió a varios facultativos, quienes le indicaron tratamiento analgésico y mejoró transitoriamente.
Hace 3 meses comenzó con un dolor intenso en dicha zona que no se alivió con opiáceos, por lo que acudió al servicio de urgencia donde se ingresó para estudio.
El examen físico fue normal, excepto el sistema osteomioarticular; se apreció la articulación de la rodilla izquierda con aumento de volumen que se extendía hasta la porción inferior del muslo con signos flogísticos inflamatorios: calor, rubor, marcada impotencia funcional e intenso dolor a la movilización que se extendía a los tejidos blandos periarticulares. No se hallaron adenopatías periféricas.
El estudio analítico fue normal, excepto la velocidad de sedimentación globular de 75 mm/1 h (VN: hasta 10 mm/1 h), la fosfatasa alcalina de 580 UI/L (VN: 40-100 UI) y la lactato deshidrogenasa 610 UI/L (VN: 130-300 UI).
La radiografía de tórax, el electrocardiograma y los ultrasonidos abdominal, urológico y retroperitoneal no mostraron alteraciones. La radiografía de rodilla izquierda posteroanterior y lateral mostró una masa ósea con aumento de las partes blandas.
No se realizó tomografía axial computarizada (TAC) del tercio distal del fémur y la rodilla izquierdas por no tener suficiente sensibilidad y especificidad en el diagnóstico de esta entidad. La TAC general no mostró metástasis ósea, retroperitoneal ni en el mediastino.
Se realizó resonancia magnética nuclear (RMN) que informó lesión osteolítica con invasión del tejido blando y numerosas imágenes con densidad negativa en su interior (Fig. 1).
Se practicó biopsia dirigida por ultrasonido, cuyo resultado reveló un TCG del tercio distal del fémur izquierdo, confirmado por estudio histopatológico (Figs. 2 y 3).
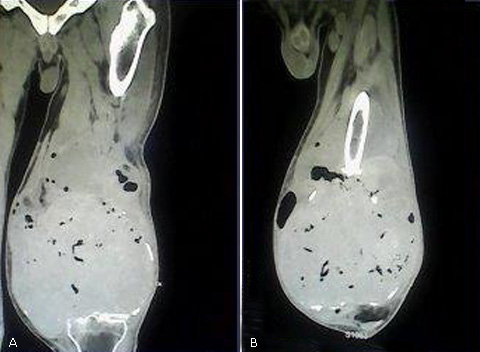
Fig. 1 Lesión osteolítica con invasión del tejido blando y numerosas imágenes con densidad negativa en su interior, correspondiente a un tumor de células gigantes.
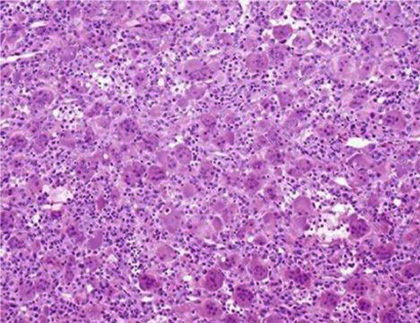
Fig. 2 Campo histológico de un área del tumor que muestra la gran celularidad y la presencia de células gigantes tipo osteoclastos. H/E 20 x.
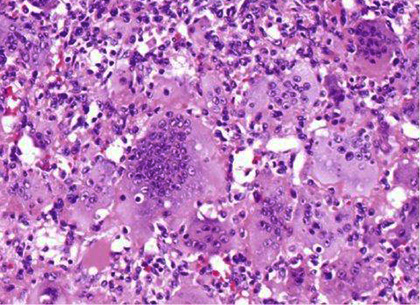
Fig. 3 Imagen histológica a un campo de mayor aumento. Obsérvese más de 30 núcleos en las células gigantes y núcleos similares redondos en las células pequeñas acompañantes. H/E 40 x.
Teniendo en cuenta las características clínico-radiológicas y la demora entre el inicio de los síntomas y el diagnóstico, se practicó la amputación del miembro afectado. Después de 10 meses de observación, el paciente se mantenía totalmente asintomático, libre de manifestaciones clínicas recurrentes o de metástasis. Fue enviado al Servicio de Oncología para valorar e tratamiento con radioterapia.
Discusión
El TCG de hueso es un tumor raro de características benignas con una evolución muy agresiva. Representa el 5 % de los tumores óseos y el 20 % de los tumores óseos benignos.3,4 La mayor frecuencia (80 %) se encuentra entre los 20-40 años y es raro después de estas edades. La incidencia general puede variar de 2-8 %; en edades pediátricas aparece en menores de 14 años (3 %).1,5 Algunos autores señalaron que afectaba con mayor frecuencia al sexo masculino.2,3,4 Este paciente era masculino y tenía 19 años, lo que corresponde con la literatura. Se señala, ademá,s que es más común en asiáticos que en caucásicos.3
Estos tumores afectan las epífisis y metáfisis de los huesos largos en los adultos, pero en los adolescentes quedan confinados proximalmente por la placa de crecimiento y afectan solo las metáfisis. La mayoría de estos tumores aparecen alrededor de la rodilla, y las localizaciones habituales son el fémur distal y la tibia proximal (50-65 %) y radio distal (10-20 %). En este caso se afectó fundamentalmente el fémur y la tibia. La localización cerca de las articulaciones hace que los pacientes sufran de dolores articulares intensos como ocurrió en este paciente, cuyo dolor solo se aliviaba con opiáceos. En ciertas oportunidades se manifiesta por fracturas patológicas y síntomas neurológicos, no reportados en este caso.
El tiempo promedio de diagnóstico entre el inicio de los síntomas y su diagnóstico varía entre 6-8 meses,2,3,4,6 pero en este caso demoró 2 años, lo cual trajo como consecuencia la terapeútica no beneficiosa para el enfermo. La mayoría de los tumores son solitarios, pero existen también formas multicéntricas, especialmente en la parte distal de los miembros.1,2,4,5,6
El origen de este tumor no está bien dilucidado, pero se piensa que los TCG se producen a partir de monocitos circulantes que tienen diferenciación osteoclástica. Las células del estroma desempeñan un papel importante en el desarrollo y la perpetuación del tumor, ya que liberan paracitocinas I de los macrófagos e IL-8 que favorecen el reclutamiento de macrófagos celulares existentes que aumentan, a su vez, la actividad de la adenilato ciclasa, la cual ofrece a estas células propiedades similares a la de los osteoclastos normales.3,6,7,8
Radiológicamente, el TCG presenta lesiones puramente líticas y excéntricas que erosionan la placa ósea subcondral, es frecuente que la cortical que cubre el tumor se destruya y aparezca una masa de tejido blando contorneado por una delgada cáscara de hueso reactivo. Las imágenes de este caso demostraron las características descritas por Enneking,4 que agrupa las lesiones radiográficas en tres estadios: el primero corresponde a un TCG latente y benigno; el segundo presenta márgenes bien definidas con una cortical adelgazada e insuflada y el tercer estadio, considerado como agresivo, consiste en lesiones sintomáticas de crecimiento rápido asociado o no a fracturas patológicas, con destrucción del hueso cortical y esponjoso medular con ruptura de la cortical e invasión de partes blandas. La RMN permite una evaluación más detellada de los componentes de la lesión (tumoración sólida de contornos fácilmente reconocibles).
El diagnóstico diferencial debe realizarse con lesiones líticas de hueso tales como sarcoma de Ewing, condroblastoma, aneurisma quístico del hueso, fibrosis no osificada y condrosarcoma.4,5,6 Desde que este tumor fue definido por Jaffé,1 como una entidad clínico-patológica con características radiográficas e histológicas, se han dividido en grados del I al III: el grado I no muestra atipia celular; en el grado II el núcleo es atípico con moderado pleomorfismo y escasas mitosis (estos cambios no son tan prominentes para considerarlos sarcoma y fue el que presentó este enfermo) y en el grado III las células estromales son francamente malignas y con capacidad de provocar metástasis. Basándose en el aspecto histológico, no es posible predecir que un TCG va a recurrir o causar metástasis, esto último ocurre en el 1-2 % de los casos y afectan a pulmones e hígado.9,10,11,12,13
El mejor tratamiento de estas lesiones neoplásicas es la resección quirúrgica total con márgenes amplios. Se han descrito recidivas hasta en 20-75 % cuando las lesiones solamente se tratan con curetaje simple. Hay referencias bibliográficas de tratamiento único con radioterapia y buen pronóstico para la vida.2
Conclusiones
El TCG es un tumor benigno infrecuente que presenta potencial de crecimiento, agresividad y recidiva. Tiene tendencia a la invasión local y destrucción ósea. La clínica está dada por dolor, aumento de volumen e impotencia funcional de las zonas óseas afectadas, y la radiografía de hueso es el primer método de estudio.
Es fundamental en estas neoplasias el periodo que transcurre desde el inicio clínico y su diagnóstico: mientras más tardío es el diagnóstico, peor es el resultado terapeútico, y aunque las metástasis son poco frecuentes, se deben considerar en el momento del diagnóstico.

