










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El monitoreo de la calidad de los recursos hídricos se ha convertido en una actividad fundamental para el hombre debido a su papel esencial en la sostenibilidad de la vida, el desarrollo y el medio ambiente. Ante la complejidad de los factores y la extensa selección de variables cruzadas que se deben considerar para describir el estatus de los cuerpos de agua en términos cuantitativos, se han establecido parámetros físicos, químicos y microbiológicos que determinan su calidad. Sobre esta base se definen diferentes clases o tipos de agua acorde a su uso, por ejemplo: agua de consumo humano, agua para actividades agrícolas, pecuarias, industriales, entre otras. La alteración o modificación de estos criterios de calidad producto de actividades socioeconómicas que limitan o perjudican su uso se define como contaminación.
Entre los agentes contaminantes más relevantes se encuentran los hidrocarburos totales de petróleo (TPH, por sus siglas en inglés); éstos pueden estar presentes en los sistemas de almacenamiento y las fuentes de abastecimiento subterráneas y superficiales debido a fugas, derrames, vertimientos y al deficiente manejo de residuos que contengan estos compuestos orgánicos. La contaminación con TPH produce un cambio en las características organolépticas del agua que induce al rechazo de los consumidores; su ingestión representa un riesgo para la salud y genera impactos negativos sobre los ecosistemas por lo que resulta imprescindible la planificación y ejecución de acciones preventivas, correctivas y de mitigación para la protección del agua (Téllez, 2016; Vammen y Vaux, 2019; Okparanma y Mouren, 2017).
El estudio de este tipo de contaminación puede alcanzar una gran complejidad, determinada por la naturaleza del contaminante, los métodos de análisis que se empleen y la manera en que se proceda en la ejecución del mismo. Existen diferentes métodos analíticos que se emplean para esos fines entre los que se encuentran los métodos cromatográficos, métodos de partición gravimétrica y métodos espectroscópicos de infrarrojo y ultravioleta, cada uno con sus ventajas y limitaciones (Álvarez et al, 2014; Santana et al,2016; Rohner, 2018). Los métodos de partición gravimétrica a pesar de ser los menos costosos presentan una baja sensibilidad; los métodos cromatográficos son altamente selectivos y sensibles, pero se consume un mayor el tiempo de análisis y se requiere el empleo de patrones de alta pureza cuyo costo es elevado; los métodos espectroscópicos son rápidos, sensibles y económicos.
La espectroscopía infrarroja de rango medio con transformada de Fourier (FTMIR, por sus siglas en inglés) se ha utilizado ampliamente para estos fines. Este método instrumental se fundamenta en la medición del área o la intensidad de las vibraciones de estiramiento y/o doblaje de los enlaces C-H presentes en los hidrocarburos. En la mayoría de los casos la forma para medir la absorción de estos
compuestos involucra solo la señal alrededor de 2930 cm-1, la cual es característica de los grupos metilenos. Existen otras metodologías en las que se consideran las señales -CH de metilos y =CH de aromáticos ubicadas en torno a 2960 cm-1 y 3030 cm-1, respectivamente. En los últimos años se ha implementado la utilización de las bandas de doblaje -CH localizadas en el intervalo de frecuencias de 1460 cm-1 a 1370 cm-1 (Pisal, 2018; Tănăselia et al, 2015; Adeniji et al, 2017; Almeida et al, 2013).
Los disolventes que se emplean en la extracción de los TPH deben cumplir el requisito fundamental de exhibir una transmisión adecuada en la región de interés además de no reaccionar y ser soluble con la muestra. Muchos de estos compuestos se prohibieron debido a los daños que han ocasionado en la capa de ozono, ejemplo el 1,2-tricloro-1,2,2-trifluoretano (freón 113) y el tetraclorometano (CCL4). Actualmente uno de los disolventes que se utiliza para esta determinación es el tetracloroetileno (C2CL4) sin embargo se ha comprobado que cuando la contaminación por hidrocarburos está en el nivel de agua potable los porcientos de recobro pueden ser inferiores a los obtenidos con otros disolventes (ASTM E1252, 2006 y ASTM D3921, 2011).
En trabajos de investigación anteriores se desarrolló un procedimiento para cuantificar los TPH a partir de datos de espectroscopía infrarroja. Los resultados obtenidos permitieron establecer el análisis de éstos contaminantes en aguas residuales (Dago-Morales A et al, 2016). El objetivo general de este trabajo es desarrollar un procedimiento analítico para la determinación de hidrocarburos totales de petróleo en matrices acuosas mediante el empleo de la espectroscopía infrarroja de rango medio y tetracloroetileno como disolvente de extracción. Para verificar que el mismo es adecuado se realizó la validación a partir de muestras en blanco fortificadas en el laboratorio y se evaluaron parámetros de desempeño analítico.
MATERIALES Y MÉTODOS
Preparación de las muestras de calibración.
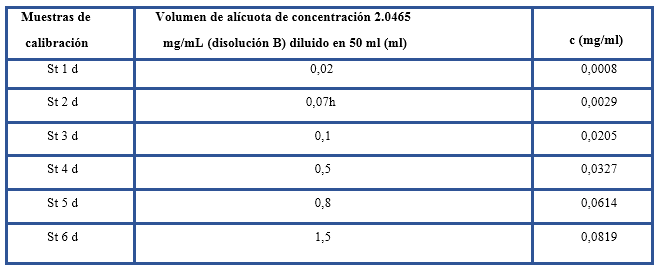
En la preparación de las muestras de calibración se utilizó una mezcla estándar de hidrocarburos: 37,5 % de Isooctano, 37,5 % de n-hexadecano y 25 % de Clorobenceno (ASTM E 1252, 2006). Se pesó 1 ml de este material y se enrasó con tetracloroetileno en un volumétrico de 100 ml (disolución A). Para considerar un intervalo más bajo de concentración se obtuvo una disolución B tomando una alícuota de 2,5 ml de A y diluyendo en un volumétrico de 10 ml con el disolvente orgánico; a partir esta disolución se ensayaron 6 muestras (tabla 1).
Registro de los datos espectrales
Los espectros infrarrojos se obtuvieron mediante la técnica de transmisión en celdas de cuarzo rectangulares (ASTM E168, 2016). Se empleó un espectrómetro FT-MIR modelo Frontier (PerkinElmer Spectrum, 2015). Las condiciones de registro fueron las siguientes:
Modo fotométrico: Absorbancia
Intervalo de frecuencia: 3200 a 2700 cm-1
Resolución: 4 cm-1
Número de barridos: 32
Porta muestra: celda de cuarzo de 30 mm de paso óptico
Previo a la colección de datos se obtuvo un espectro del fondo con el disolvente y luego se realizó un escaneo de simple haz para cada celda, también con el disolvente, con propósito de sustraer la contribución del mismo (Smith, 2011).
Modelo de Regresión.
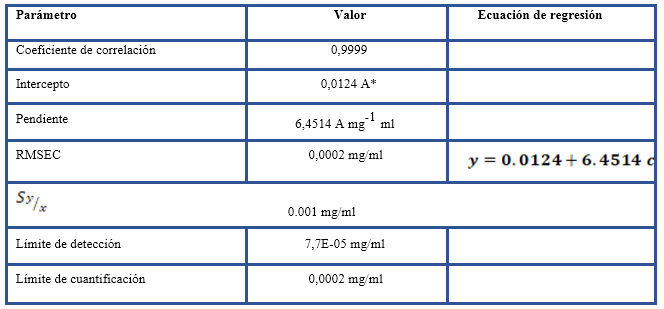
A partir de los datos espectrales del conjunto de las muestras de calibración se calculó el modelo de regresión. La medición de la altura de la señal correspondiente a la vibración de estiramiento C-H asimétrico de metilenos (~2930 cm-1) se efectuó a partir de valores establecidos de las dos bases de la señal: 3200 cm-1 (base 1) y 2700 cm-1 (base 2) y de valores iniciales y finales de 2944,70 cm-1 y 2881,25 cm-1. El ajuste de los puntos se realizó con un polinomio de grado 1. Posterior al proceso de calibración se obtuvo la ecuación de regresión, así como las concentraciones estimadas a partir del modelo. Los cálculos se realizaron con el programa PerkinElmer Spectrum Quant, 2015.
Para efectuar la evaluación del desempeño analítico se consideró el coeficiente de correlación (r), el error estándar de calibración (RMSEC, por sus siglas en inglés) y los gráficos de áreas vs concentración teórica, concentración teórica vs concentración estimada por el modelo y concentración teórica vs concentración residual (Olivieri, 2017; Miller & Miller, 2010, Magnusson and U. Örnemark, 2016). Además, se calcularon los límites de detección (LD) y de cuantificación (LC) a partir de la desviación estándar de los residuales de la regresión (ICH Harmonised Tripartite Guideline, 2005).
Validación del modelo.
En la validación de los modelos se empleó el método de añadido recobrado, para lo cual se utilizaron blancos fortificados en el laboratorio (muestra control). Se ensayaron 2 niveles de concentración por triplicado cercanos al límite de cuantificación. La muestra control de concentración 0,88 mg/l se preparó en el laboratorio a partir de combustible diésel sin aditivar. Los 6 volúmenes se añadieron de manera independiente a 6 litros de agua destilada tipo II y se ajustó el pH hasta 3 con HCL 1:1. El proceso de extracción se realizó utilizando tetracloroetileno grado espectroscópico en un embudo separador provisto de llave y tapa de teflón. Se efectuaron tres extracciones con 15 ml del solvente. Los extractos se filtraron por sulfato de sodio anhidro y se enrasaron en volumétricos de 50 ml. Por último, se agregaron 1,5 g de sílica gel a cada volumétrico para remover los compuestos polares (APHA- AWWA-WEF, 2017).
El procedimiento utilizado en el registro de los espectros infrarrojos se realizó acorde a lo descrito en el apartado Registro de datos espectrales con la particularidad de que para obtener el espectro de fondo en lugar del solvente se utilizó un blanco reactivo de agua destilada el cual se sometió al mismo proceso de extracción que las muestras de validación.
Las concentraciones estimadas se obtuvieron en mg/ml. El reporte de éstos resultados se realizó mg/l para lo cual se empleó la ecuación siguiente:
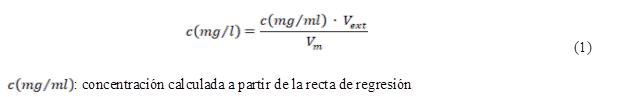
Para efectuar la evaluación del desempeño del modelo en esta etapa se consideró el por ciento de recobro y la desviación estándar relativa para cada nivel de concentración. Además se calculó el error asociado a las concentraciones para los niveles antes mencionados (desviación estándar del error global en la concentración) y se reportó como límite de confianza de acuerdo a:  ±t(n-2) *
±t(n-2) *  (Thompson, 2006; Olivieri, 2014).
(Thompson, 2006; Olivieri, 2014).
ESTADÍGRAFOS UTILIZADOS EN LA EVALUACIÓN DE LOS RESULTADOS.
Etapa de calibración
1. Raíz cuadrada del error medio de calibración (RMSEC, por sus siglas en inglés).

Donde:
xi |
: concentraciones de referencia |
xest |
: concentraciones estimadas a partir del modelo |
n |
: número de muestras utilizadas en la calibración |
2. Coeficiente de correlación (r).
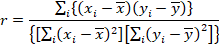 (3)
(3)Donde:
|
: media de xi |
|
: alturas utilizadas en la regresión |
|
: media de |
|
: alturas de la señal utilizadas para obtener la recta de calibración |
|
: alturas de la señal utilizadas obtenidas al evaluar xi en la ecuación de la recta de calibración |
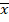
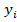
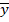
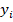
3. Límite de detección (LD).
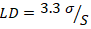 (4)
(4)4. Límite de cuantificación (LC).
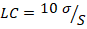 (5)
(5)5. Desviación estándar de los errores aleatorios en la dirección y (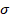 ).
).
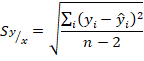 (6)
(6)Etapa de validación
6. Desviación estándar del error global en la concentración (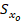 ).
).
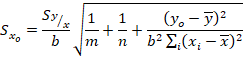 (7)
(7)Dónde:
m |
: número de réplicas |
n |
: número de muestras utilizadas en validación |
|
: promedio de las alturas de la señal utilizadas para las m réplicas |
7. Porciento de recobro.
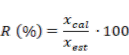 (8)
(8)8. Desviación estándar relativa (DER)
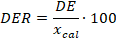 (9)
(9)RESULTADOS Y DISCUSIÓN
Modelo de regresión
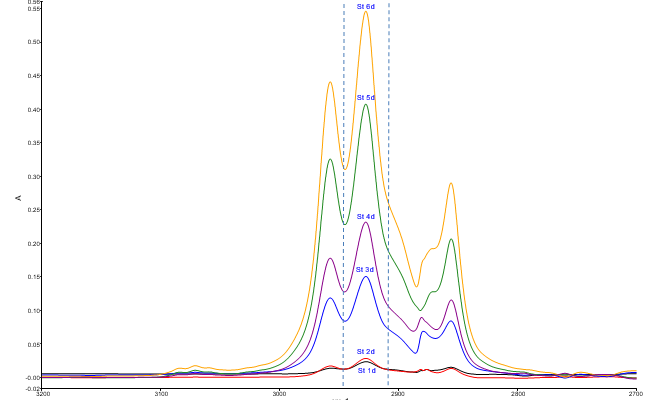
En la figura 1 se presentan los espectros infrarrojos de las muestras de calibración. Entre líneas discontinuas se destaca el intervalo de frecuencias que se empleó para el cálculo de la altura máxima de la señal en el entorno de 2930 cm-1 característica de la vibración de estiramiento C-H de compuestos alifáticos. Se observa un incremento proporcional de la absorbancia con la concentración. El límite de cuantificación que se obtuvo se corresponde con lo que refieren las normas internacionales respecto a la cantidad máxima permisible de hidrocarburos en fuentes de abastecimiento de agua (tabla 2). El resto de los parámetros de desempeño que se evaluaron y la ecuación de regresión se consideraron conformes para el fin previsto (Olivieri, 2014; Standards and Guidelines for Contaminants in Massachusetts Drinking Waters, 2018).
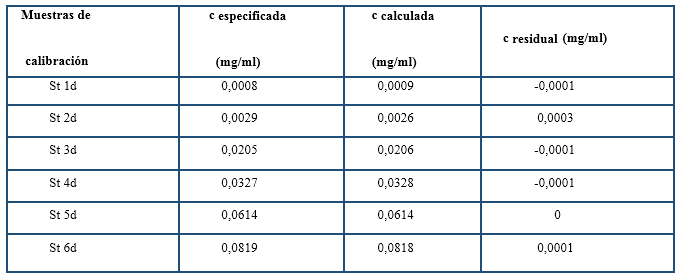
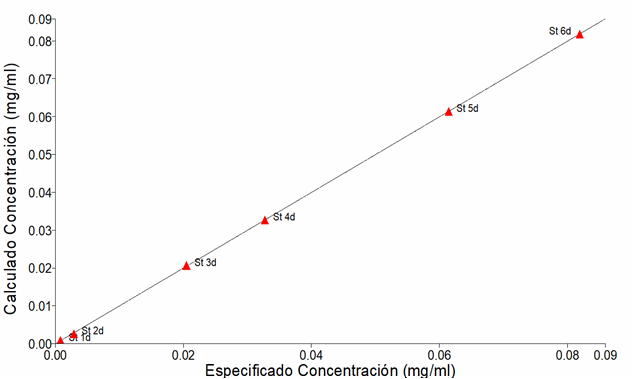
En la figura 2 se observa un buen ajuste lineal entre las concentraciones calculadas y especificadas lo cual determinó el alto coeficiente de correlación (Olivieri, 2017; Miller & Miller, 2010). La mayor concentración residual fue de 3 x 10-4 mg/ml que corresponde al St 3d (tabla 3).
El error global de la concentración () fue de 0,0001 mg/l el cual es adecuado para los bajos niveles de concentración ensayados (Magnusson and U. Örnemark, 2016). Los límites de confianza se reportan a continuación:
En la tabla 4 se presentan otras características de desempeño del procedimiento analítico. La desviación estándar relativa (DER) en todos los casos fue inferior al 5 % por lo que puede plantearse que el método desarrollado es preciso (Olivieri, 2017). Los valores de recobrado por otra parte, en 1 de los 2 niveles ensayados estuvo en el intervalo entre 80 y 120 % lo cual, de acuerdo a la literatura especializada, se considera un resultado satisfactorio. En el caso de la concentración de referencia0,264 mg/L, este parámetro no superó el límite inferior del criterio de aceptación lo cual se puede atribuir a pérdidas por solubilidad en el procedimiento de extracción; no obstante, si se tiene en cuenta la baja concentración y que la diferencia entre el valor calculado y el de referencia es del 3 %, se puede concluir que la metodología analítica que se empleó es eficiente en las condiciones de trabajo establecidas (Standards and Guidelines for Contaminants in Massachusetts Drinking Waters, 2018; Olivieri, 2011, 2014, 2017).

CONCLUSIONES
Se desarrolló un procedimiento analítico para cuantificar hidrocarburos totales del petróleo en agua por espectroscopía infrarroja de rango medio en el intervalo de concentraciones de 0,0002 mg/ml a 0,0819 mg/ml. La validación del método de regresión se realizó a partir de muestras en blanco fortificadas en el laboratorio cuyos valores de concentración se establecieron próximos al límite cuantificación: 0,2640 mg/l y 0,5280 mg/l. Las desviaciones estándares relativas fueron inferiores al 5 % en ambos casos y los recobrados de 77 % y 84 % los cuales se consideran adecuados. El procedimiento propuesto puede emplearse para determinar el contenido de hidrocarburos en fuentes de abastecimiento de agua.

