










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Ciencias Médicas de Pinar del Río
versión On-line ISSN 1561-3194
Rev Ciencias Médicas vol.21 no.4 Pinar del Río ago. 2017
ARTÍCULO DE REVISIÓN
Caracterización clínica y manejo del Síndrome de Ehlers Danlos
Clinical characterization and management of the Ehlers-Danlos syndrome
Marianela Puerto Martínez1
1 Médica. Especialista de Primer Grado en Pediatría. Policlínico Universitario Manuel Pity Fajardo. Guane. Cuba. yailincalero@infomed.cu
Recibido: 28 de marzo de 2017
Aprobado: 23 de junio de 2017
RESUMEN
Introducción: el síndrome de Ehlers Danlos es una enfermedad poco frecuente cuyo diagnóstico es en muchos casos subvalorado. Es una enfermedad hereditaria, producida por mutaciones cromosómicas, que pueden llegar a tener comportamiento autosómico dominante, recesivo o ligado al cromosoma X.
Objetivo: se clasifica en 6 subtipos, siendo los más frecuentes el tipo I y III, y el más grave de todos, el subtipo vascular. Es de importancia el realizar un diagnóstico clínico apropiado.
Desarrollo: esta enfermedad se caracteriza principalmente por defectos en la producción del colágeno, resultado del daño presente en las enzimas que se encargan de la síntesis del mismo. La variedad con la que se presenta, hace que se considere heterogénea, por lo que se destacan grandes tipos de este padecimiento, cada uno de ellos con signos y síntomas similares, pero con progresión y evolución distinta, presentando en común, la hiperlaxitud articular, hiperlaxitud articular y la hiperequimosis por fragilidad de los vasos sanguíneos. Actualmente no existe ningún tratamiento específico para la misma.
Conclusiones: se requieren estudios genéticos y bioquímicos que ayuden a identificar los defectos coexistentes. Algunas formas del síndrome son comunes, mientras que otras muy raras. Es importante realizar la exploración física completa y buscar las características específicas de cada subtipo para identificarlo con la mayor precisión posible.
DeSC:SÍNDROME DE EHLERS-DANLOS; FIBROMIALGIA; INESTABILIDAD DE LA ARTICULACIÓN; CROMOSOMA X;TEJIDO CONECTIVO.
ABSTRACT
Introduction: Ehlers-Danlos syndromeis a rare disease which diagnosis is in many cases underestimated. It is an inherited disease, caused by chromosomal mutations, which may have autosomal dominant, recessive or X-linked behavior.
Objective: it is classified into 6 subtypes, being the most frequent type I and III, and the most serious of all, is the vascular subtype. It is important to make an appropriate clinical diagnosis.
Development: this disease is characterized mainly by defects in the production of collagen, and as a result of the present damage in the enzymes responsible for its synthesis. The variety with which it is presented causes it to be considered heterogeneous; it is why there are great types of this disease, each with similar signs and symptoms, but with different progression and evolution, presenting joint hyperlaxity, hyperelasticityof the skin and hyper-ecchymosesdue to fragility of the blood vessels. At present there is no specific treatment for it.
Conclusion: genetic and biochemical studies are needed to help to identify the coexisting defects. Some forms of the syndrome are common, while others are very rare. It is important to perform the complete physical examination and look for the specific characteristics of each subtype to identify it with the highest possible precision.
DESC: EHLERS-DANLOS SYNDROME; FIBROMYALGIA; JOINT INSTABILITY X CHROMOSOME; CONNECTIVE TISSUE.
INTRODUCCIÓN
La primera descripción del síndrome data de 1682 y se la debemos a Job Von Meekeren (1611-1666) un cirujano de Ámsterdam. 1
El siguiente informe apareció en Moscú en 1891, donde el Dr. A. N. Tschernogobow presentó dos casos (un niño de 17 años y una mujer de 50). La aportación más importante de este autor fue que asoció las múltiples manifestaciones de estas personas, y dijo que eran debidas a un trastorno generalizado del tejido conectivo. A él corresponde el mérito de la descripción clásica del síndrome que en realidad debería conocerse por su nombre. 2
Posteriormente en París (1899) Edward Ehlers presentó otro caso en una reunión de la Sociedad de Veneorología y Dermatología. El enfermo tenía hiperlaxitud articular y variados y múltiples problemas ortopédicos. Tenía la piel hiperextensible y había desarrollado lesiones pigmentadas sobre las prominencias óseas debido a traumatismos mínimos y también presentaba debido a los mismos una tendencia hemorrágica relativamente importante. 3
Nueve años más tarde, en 1908 Henri-Alexandre Danlos, expuso otro caso. Afirmó que las lesiones sobre las prominencias óseas eran postraumáticas y que el paciente tenía un defecto inherente, que él denominó "cutis laxa".
Frederich Parkes-Weber en un artículo publicado en el Diario de la Sociedad Británica de Dermatología propuso el nombre de síndrome de Elhers Danlos para denominar a esta enfermedad. Este nombre ganó aceptación entre los miembros. 4
El síndrome de Ehlers Danlos es un grupo heterogéneo de trastornos hereditarios del tejido conectivo caracterizado por síntesis de colágeno anormal que afecta la piel, ligamentos, articulaciones, vasos sanguíneos y otros órganos causado por mutaciones en los genes que codifican el colágeno fibrilar tipo I, III y V o enzimas comprometidas en la modificación post-translacional de dichos colágenos. 5
Con el objetivo de unificar criterios diagnósticos y mejorar la atención integral y la expectativa de vida de estos enfermos, en 1997 se realizó la última clasificación del síndrome por un grupo de expertos en Villafrance que fue publicada al año siguiente e incluye 6 grupos principales, siendo los más frecuentes el I y el III y el más grave el tipo vascular debido a las muertes tempranas por rupturas arteriales. El tipo cifiescoliótico (VI), la artrocalasia (VII-A y B) y la dermatosparaxis son los menos frecuentes. Cada uno de estas variantes posee signos clínicos mayores y menores que definen sus características particulares. 6
La hipermovilidad articular, la extensibilidad y la hiperlaxitud como su forma extrema pueden producir daños articulares de diversa severidad. Junto a ellas, la fragilidad cutánea y la debilidad muscular fueron desde un inicio los elementos que distinguieron la clínica de estos pacientes. Posteriormente las descripciones más detalladas y la disponibilidad de estudios bioquímicos y genéticos han permitido describir diferentes mutaciones relacionadas con una expresión clínica variable dentro de cada tipo de esta entidad. 7
Aproximadamente un 10% de la población adulta es hiperlaxa. La prevalencia varía entre los diversos grupos étnicos y es mayor entre las mujeres. Sin embargo, el síndrome de hiperlaxitud (hiperlaxitud + síntomas) afecta solamente a una minoría de las personas hiperlaxas. 8
Hoy en día, la mayoría de los especialistas que estudian estas patologías consideran que el síndrome de hiperlaxitud articular y el síndrome Ehlers-Danlos tipo hiperlaxitud (denominado anteriormente tipo III) quizás sean el mismo proceso. 9
El síndrome de hiperlaxitud articular es "una enfermedad invisible", es decir, las personas que lo padecen tienen una apariencia normal y debido a esto puede parecer que están perfectamente bien, pero a menudo el dolor severo y la limitación funcional los acompañan cada día. Debido a que los tejidos conectivos son muy frágiles, la probabilidad de padecer cualquier tipo de lesión (luxaciones, fracturas, esguinces, tendinitis) aumenta, incluso cuando realizamos las actividades más ligeras de la vida diaria. Actividades cotidianas como bañarse, subir y bajar escaleras, escribir, utilizar el ordenador, la preparación de las comidas, especialmente cortar vegetales, pueden resultar difíciles en aquellos pacientes que presentan un mayor grado de afectación.
Un aspecto que no suele usualmente tenerse en cuenta en las consultas médicas y que contribuye al sub-diagnóstico de la enfermedad es que, con la edad, la hiperlaxitud disminuye, pero los síntomas y otras complicaciones resultantes de la hiperlaxitud suelen aumentar. Entre estas complicaciones podemos destacar la osteoartritis secundaria, la osteoporosis con las fracturas resultantes, y la pérdida del equilibrio particularmente en personas mayores, que provocará caídas. 10 Incluso si una sola articulación hiperlaxa causa dolor o inestabilidad el diagnóstico debe seguir siendo síndrome de hiperlaxitud articular.
La prevalencia de este trastorno es difícil de establecer debido ya que se han utilizado diferentes criterios diagnósticos y porque su frecuencia varía dependiendo de la raza, edad y sexo. Existiría en 10% a 15% de la población occidental. Su frecuencia es mayor en los asiáticos y negros, y en éstos, más que en los caucásicos. Es más frecuente en mujeres y niños. En la mayoría de los casos el compromiso es pauciarticular, lo que dificulta el diagnóstico. 11-14
El síndrome de hiperlaxitud articular es relativamente frecuente en nuestra población de niños escolares sanos y se puede asociar con algunas manifestaciones clínicas como escoliosis, pies planos, dolor lumbar, piel fina y párpados caídos.
Un adecuado conocimiento de esta entidad entre los pediatras y médicos será de utilidad para evitar errores diagnósticos, tomar medidas preventivas adecuadas para que no se agrave el cuadro clínico y no crear, ni falsas expectativas ni ansiedad extrema, en pacientes y familiares, que al no conocer exactamente los detalles de esta entidad, ni su relativa frecuencia en la población, recurren a las diferentes especialidades médicas maximizando el problema o temerosos de posibles complicaciones.
Se hace necesario la atención particularizada de cada paciente, con un exhaustivo interrogatorio y examen físico, para determinar si es necesario interconsultar con otras especialidades médicas, lo que se haría en dependencia de las manifestaciones clínicas de cada paciente, por lo que consideramos una práctica errónea el hecho de que a los pacientes se les indique una "batería de interconsultas" similar para todos, que lejos de resolver el problema, crea ansiedad, recarga las consultas externas y en la mayoría de los casos resultan innecesarias por no encontrar manifestaciones o alteraciones propias de esa especialidad.
El tener hiperlaxitud articular sin síntomas es sólo una particularidad, pero si hay síntomas debidos a su condición se trata de una enfermedad, denominada síndrome de hiperlaxitud articular. Se representa el extremo superior de una curva de distribución de Gauss del rango de movimiento articular normal. Su prevalencia en niños sanos varía entre 5-12% de la población occidental, pudiendo alcanzar hasta el 25% en otras regiones del planeta.
El síndrome de hipermovilidad articular se observa con mayor frecuencia en mujeres que en hombres, es dependiente de la edad hasta desaparecer prácticamente en edades tardías. Esta entidad ha sido asociada a dolores musculo esqueléticos en la infancia, a osteoartrosis precoz y a rupturas ligamentarias. De acuerdo a los criterios usados para definirla, la hiperlaxitud articular puede ser localizada o generalizada, heredada o adquirida y es notablemente influenciada por la edad, sexo y raza. Para la mayoría de los médicos y de la población se trata de una curiosidad o "un juego de niños" o "un acto circense" y no de un problema médico potencialmente serio. 8
Las razones de esta falta de interés son diversas. A los pacientes se les diagnostican problemas puntuales como tendinitis, bursitis o subluxaciones, sin identificar que padecen de un cuadro más complejo. Si se reconoce que el enfermo tiene hiperlaxitud, no se presta atención a su significado ni a las posibles complicaciones. La muy alta frecuencia de hipermovilidad articular, la falta de conocimiento de que ella está relacionada a daño de múltiples órganos, unido al hecho de que su cuadro clínico no es dramático, que no existen signos inflamatorios acompañantes y que no están disponibles exámenes de laboratorio ni signos radiológicos típicos asociado a la sensación de que el tratamiento no es efectivo, hace que los médicos no se interesen en el diagnóstico de estas afecciones. A los estudiantes de Medicina no se les insiste curricularmente lo suficiente sobre esta enfermedad e incluso la observación en la práctica del trabajo de los médicos en general, incluidos los reumatólogos, evidencia que no han sido entrenados en cómo examinar al enfermo hiperlaxo y la mayoría de ellos no conocen o no utilizan los criterios diagnósticos que permiten reconocer a estos pacientes. Disponer de hojas impresas con los criterios de Beighton para definir el compromiso articular y los aspectos clínicos incluidos en la clasificación de Villefranche del síndrome de Ehlers-Danlos, así como su correcta y sistemática utilización en los enfermos para comprobar si cumplen los mismos, facilitará la orientación clínica frente a las diferentes variantes de esta patología.
Genética
El subtipo clásico es el más frecuente, pues corresponde a 35% de los pacientes con síndrome de Ehlers-Danlos. Se hereda de forma autosómica dominante a través de mutaciones en los genes COL5A1 o COL5A2. Las mutaciones de novo son comunes, ya sea por translocaciones o sustitución de cisteína en los genes implicados que codifican para las cadenas de procolágeno, proa1 (V) o proa2 (V), respectivamente, que producen anormalidades en el colágeno V.
El subtipo de hipermovilidad, el segundo en frecuencia, también cuenta con herencia autosómica dominante, pero su expresión es variable. No se conocen las alteraciones genéticas o moleculares. La forma vascular (tipo IV) su prevalencia estimada es de 1:100,000 individuos. Es de herencia autosómica dominante. El gen COL3A1 codifica la cadena de procolágeno proa1 (III). Se ha reportado que la sustitución del último nucleótido del exón 34 lleva un salto que altera la síntesis, secreción y estructura del colágeno tipo III. La artrocalasia es de herencia autosómica dominante y sus alteraciones ocurren en los genes COLIA1 o COLIA2. Las variaciones bioquímicas consisten en mal acoplamiento del propéptido aminoterminal del colágeno tipo I y en las cadenas de procolágeno proa 1 o proa2.Los subtipos restantes (cifoescoliosis y dermatosparaxis) son los únicos de herencia recesiva. En el de cifoescoliosis, la mutación del gen PLOD1 se encuentra en el cromosoma 1p36.2; ocasiona deficiencia de la enzima lisil-hidroxilasa y disminuye la hidroxilación de procolágeno a colágeno I y III. El subtipo de dermatosparaxis es el menos frecuente, sus principales alteraciones se producen en el gen ADAMTS, donde los defectos bioquímicos ocurren en el propéptido aminoterminal del colágeno tipo I, ocasionado por la mutación del gen de la N-proteinasa de procolágeno I. 15
Modo de herencia
El síndrome de Ehlers Danlos se hereda en la mayor parte de sus variantes con un patrón de herencia autosómica dominante.
Riesgos en los miembros de la familia con síndrome de Elhers Danlos tipo hiperlaxitud:
Padres de una persona afectada
-
La mayor parte de las personas diagnosticadas con síndrome tipo hiperlaxitud tienen un padre afecto, aunque generalmente es necesaria una historia cuidadosa y un examen de los padres para reconocer que, en ausencia de complicaciones serias, uno de los padres (y a veces los dos) tienen una historia de laxitud articular, facilidad para la equimosis y piel suave.
-
Una persona con el tipo hiperlaxitud puede tener la enfermedad como resultado de una mutación de novo; la proporción de casos causados por una mutación de novo es desconocida.
-
Las recomendaciones para evaluación de los padres de una persona afectada con mutación aparentemente de novo incluye una historia cuidadosa y un examen buscando historia de laxitud articular, facilidad para la equimosis y piel suave.
Hermanos de la persona afectada
-
El riesgo en los hermanos de la persona afectada depende del estatus genético de los padres de la persona afectada.
-
Si uno de los padres de la persona afectada también está afectado, el riesgo que uno de los hermanos padezca la enfermedad es de un 50%.
-
Cuando los padres no están afectados clínicamente, el riesgo de los hermanos de padecer la enfermedad parece ser muy bajo.
Descendencia de la persona afectada
Cada hijo de una persona afectada por el síndrome tipo hiperlaxitud tiene un 50%de posibilidades de heredar la mutación. De todas maneras, debido a la marcada variabilidad clínica, es difícil predecir la severidad entre los futuros afectados.
Otros miembros de la familia de la persona afectada
El riesgo de otros miembros de la familia depende del estatus genético de los padres de la persona afectada. Si uno de los padres está afectado, los miembros de su familia tienen, asimismo, riesgo de estarlo.
Temas relacionados con el consejo genético
Debido a que es probable que la metodología de las pruebas genéticas y nuestra comprensión de los genes, mutaciones y enfermedades se mejorarán en el futuro, se ha decidido crear los bancos de ADN que son lugares donde se almacena el ADN (extraído generalmente de leucocitos en sangre), para un uso futuro. 16
Pruebas prenatales
No existen pruebas prenatales que ayuden al diagnóstico de esta entidad.
Consideramos que el mayor conocimiento de estas enfermedades por el público y por los médicos redundará en una mejor calidad de vida de los enfermos, facilitará el diagnóstico y mejorará la prevención y tratamiento precoz de las complicaciones, en especial de la osteoporosis, disautonomía y las rupturas arteriales y de órganos.
La presente monografía es un texto de consulta en esta materia para estudiantes, médicos de la atención primaria y especialistas en Ortopedia, Fisiatría, Genética y otras disciplinas. Una pormenorizada revisión del contenido científico de ésta permite apreciar con objetividad el momento en el que se encuentran los conocimientos acerca de este tema y contribuir en la preparación de cada una de las especialidades, lo que contribuirá a que esta obra cumpla su objetivo.
DESARROLLO
La enfermedad es habitualmente diagnosticada en niños pequeños, pre-escolares y escolares. Se aprecia retraso en la sedestación y en la marcha, separación amplia de los ojos y amplio puente nasal más marcada en el tipo IV. Habitualmente existe una historia familiar. Los pacientes con el síndrome de Ehlers-Danlos suelen tener una constitución normal. No se han descrito de forma clara alteraciones en la talla ni en la proporción de los miembros. 17
Los motivos de consulta o ingreso más frecuentes en los pacientes con este síndrome son:
— Enfermedades neurológicas: 55,3%.
— Enfermedades respiratorias: 9,3%.
— Cuadros hemorrágicos: 7,4%.
— Enfermedades diarreicas: 7,4%.
— Enfermedades dermatológicas: 5,3%.
Los hallazgos clínicos más frecuentes son:
— Hipermovilidad articular: 84,1%.
— Hiperelasticidad de la piel: 61,8%.
— Manifestaciones hemorrágicas: 44,1%.
— Fragilidad en el tejido conectivo: 36%.
Resulta llamativo encontrar un 23,6% de alteraciones neurológicas. Entre ellas, epilepsia, migraña, convulsión febril, convulsión neonatal y retraso psicomotor.
Hagamos un recorrido por los aspectos clínicos más sobresalientes:
Hiperelasticidad y fragilidad de la piel
La piel en el síndrome es hiperelástica, dejándose estirar varios centímetros para volver a su posición inicial al soltarla. La expresión cutánea de la elasticidad cutánea varía en los diferentes tipos del síndrome.
Las lesiones en la piel ayudan al diagnóstico precoz de la enfermedad. Con el inicio de la deambulación del niño y los pequeños traumatismos subsecuentes aparecen heridas profundas en codos, rodillas v cara anterior de piernas. Estas lesiones son relativamente indoloras; los márgenes tienden a retraerse y curar despacio. Las cicatrices resultantes son de características especiales; en algunos casos pueden tener aspecto papiráceo, en otros son atróficas, anchas, delgadas, con arrugas en su interior brillantes y con frecuencia pigmentadas, probablemente debido a la organización de hematomas intradérmicos o subdérmicos. En el tipo IV (vascular) la piel es tan fina que deja transparentar la red venosa subdérmica. 18
Hiperlaxitud, hipermovilidad y luxaciones articulares
La hipermovilidad articular es la manifestación más frecuentemente encontrada en la enfermedad, siendo secundaria a la hiperlaxitud articular generalizada.
Para su diagnóstico se ha utilizado el método de Carter y Wilkinson, en el que 3 o más de los siguientes criterios deben cumplirse:
1. Aposición pasiva del pulgar a la cara flexora del antebrazo.
2. Hiperextensión pasiva de los dedos hasta que se sitúan paralelos a la cara dorsal del antebrazo.
3. Hiperextensión del codo de más de 10°.
4. Hiperextensión de rodillas de más de 10°.
5. Excesivo grado de movilidad pasiva con la dorsiflexión del tobillo y eversión del pie.
La mayoría de los pacientes con la enfermedad de Ehlers-Danlos tienen una puntuación al menos de 3 en esta escala.
La laxitud afecta especialmente a los dedos de manos y pies. En casos extremos la inestabilidad en los dedos de las manos imposibilitaba a los pacientes a realizar actividades cotidianas como conectar interruptores o desenroscar tapones. La inestabilidad de rodilla y tobillo son las más frecuentes. Las caderas son hiperextendidas durante la carga para contrarrestar el genu recurvatum, permitiendo de este modo a la pelvis mantenerse en equilibrio con respecto a los pies. 33
Anormalidades de la caja torácica
El esternón deprimido se encuentra con relativa frecuencia. También pueden verse prominencias de una o varias articulaciones condrocostales. Otras veces asimetrías torácicas de menor grado, a veces asociadas a escoliosis torácicas.
Ocasionalmente, el cuello es excesivamente largo llamando la atención la verticalización pronunciada de las primeras costillas en las radiografías de tórax.
Deformidades raquídeas
La escoliosis es relativamente frecuente. Parece presentarse en cualquier variante del síndrome, pero es más frecuente en el tipo III y más severa cuando existe alteración del colágeno tipo IV. Con frecuencia no es posible controlar la deformidad con corsés. Este deterioro parece obedecer a la incompetencia de los ligamentos espinales laxos para mantener un balance raquídeo adecuado en presencia de un rápido crecimiento longitudinal. Se ha descrito también deformidad en cifosis, con acuñamiento anterior de los cuerpos vertebrales. Cuando es necesaria la cirugía raquídea en el síndrome para corregir deformidades es posible llevarla a cabo, pero se debe realizar siempre una hemostasia cuidadosa por el riesgo de hemorragia y hematomas y extremando el cuidado de las partes blandas para evitar problemas de cicatrización.
Otras anomalías esqueléticas
Se encuentran problemas esqueléticos en un 38,7% de los casos, incluyendo las deformidades raquídeas. Entre ellos, tibia vara, genu recurvatum, genu valgo, luxaciones recurrentes y luxación congénita de cadera. El pie plano se encuentra con relativa frecuencia en la enfermedad. La mayoría de estas alteraciones se observan en pacientes con índices de hipermovilidad de 3 o más, y en los casos más severos coinciden con índices de 5. No se ha descrito aumento de incidencia fracturas ni de falta de consolidación ósea. 18La artrosis parece afectar con mayor frecuencia a pacientes con este síndrome, siendo su incidencia e intensidad proporcional al grado de hipermovilidad. Otro hallazgo frecuente es la osteoporosis generalizada de las manos o de las falanges distales. 19
Calambres musculares
Se han descrito calambres musculares con relativa frecuencia y con tendencia a la desaparición en la edad adulta. Se relacionan también con el grado de hipermovilidad articular.
Tumores cutáneos
Sobre puntos de presión como codos y rodillas pueden aparecer tumores en la piel y tejido celular subcutáneo, denominados pseudotumores moluscoides, que pueden calcificarse. Estos tumores son ovoideos, con un tamaño que oscila entre 4 y 8 mm, con una estructura de densidad homogéneamente calcificada y limitada por una zona periférica de calcificación más densa a modo de "cáscara". Para algunos autores el hallazgo de estas lesiones es casi patognomónico del síndrome. Su patogenia es desconocida, aunque se sugiere ya un origen traumático, congénito o incluso secundario a la calcificación de los hematomas resultantes de la fragilidad capilar.
Diátesis hemorrágica
Las manifestaciones hemorrágicas se encuentran en tercer lugar de frecuencia en el síndrome, por detrás de la hipermovilidad de las articulaciones y de la hiperelasticidad de la piel. En algunos pacientes aparece con facilidad de equimosis y hematomas subdérmicos y en planos tisulares profundos. Su localización son los puntos de presión, manifestándose como tumoraciones de gran tamaño en nudillos, codos y rótulas.
Pueden aparecer hemorragias espontáneas, especialmente en muslos y pantorrillas, que incluso pueden requerir transfusiones sanguíneas o drenajes quirúrgicos. Si están presentes se prestará atención principal al posible desencadenamiento de síndromes compartimentales.
Según el concepto clásico de la enfermedad, la tendencia hemorrágica en el síndrome de Ehlers-Danlos es más frecuente y severa en el tipo IV, y es dependiente de la fragilidad vascular, el adelgazamiento de la piel y la ruptura de grandes arterias, más que por defectos en los factores de la coagulación o la función de las plaquetas. 20No obstante, se han reportado alteraciones de la hemostasia, en particular las anomalías plaquetarias, en casos aislados del síndrome, que en ocasiones se han considerado como un trastorno que lo acompaña. El estudio de función plaquetaria no había sido evaluado suficientemente para determinar si existen anomalías que participen en las manifestaciones de la entidad, sin embargo, la tendencia hemorrágica de diverso grado observada en los pacientes con síndrome de Ehlers-Danlos tipo III hizo necesaria su evaluación y en un estudio realizado en Pinar del Río hace tres años 15fue identificada la existencia de trastornos de la agregación y función plaquetaria como un componente de la enfermedad y el comportamiento clínico de dicha alteración consistió en la existencia predominante de sangrado cutáneo, tanto espontáneo como ante pequeños traumas. Los resultados obtenidos evidenciaron que el hallazgo de petequias y equímosis es relevante en los pacientes con síndrome de Ehlers-Danlos tipo III y que los sangramientos mucosos, aunque menos frecuentes, son también un problema clínico que puede estar presente en estos enfermos. En la investigación realizada se demostró que la mitad de niños que presentaban sangramiento cutáneo-mucoso utilizaba de forma continua medicamentos antihistamínicos (ketotifeno), lo que indica que el empleo sistemático de este fármaco puede constituir un factor de riesgo para su aparición.
Afectación ocular
Puede aparecer ectopia de cristalino, aunque es menos frecuente que en el síndrome de Marfan. La piel sobre los ojos puede ser redundante y la esclerótica con frecuencia es azulada. 21
Fenómenos circulatorios periféricos
Ocasionalmente se ha visto en este síndrome el fenómeno de Raynaud, cursando a veces con marcada osteólisis de las falanges distales de ambas manos y también acrocianosis. 22
Embarazo y parto
Se ha descrito algún caso de inestabilidad de hombro durante el embarazo, así como dolor y parestesias en brazos y manos. No son raros los problemas en la sínfisis del pubis, que es forzada durante el parto. A la laxitud propia de estos pacientes se añaden las condiciones de facilitación provocadas por un ambiente rico en «relaxina» en el intento de modificar el canal del parto que se puede traducir en dificultades para la marcha durante varias semanas del postparto por dolor sinfisario. Existe una mayor incidencia de partos prematuros y rotura prematura de membranas en estos pacientes.
Durante el embarazo y parto éstas parecen tener mayor riesgo de sufrir complicaciones vasculares. 23
Alteraciones psicológicas
La tercera parte de estos tienen cuadros de ansiedad, depresión, actitud colérica y problemas interpersonales. Los problemas psicológicos están en relación directa con el dolor crónico y discapacidad, ostracismo, dificultades sexuales y reproductivas y relaciones sociales. El tipo I de la enfermedad se relaciona especialmente con dolor y problemas psicológicos. 24
Características de la enfermedad en Pinar del Río
En una investigación sobre la caracterización del tipo III del síndrome (hipermovilidad articular generalizada) en la población pediátrica de la provincia de Pinar del Río publicada en el año 2013 4, no se evidenció predominio de género como ha sido señalado en la literatura sobre el tema 46probablemente por el hecho de que las series son de menor número de pacientes y que incluyen niños y adultos.
Los antecedentes familiares fueron predominantemente en la línea materna y fue inusual en ambos progenitores asociados. A pesar de la herencia autosómica dominante, la ausencia de antecedentes familiares conocidos en un grupo de los pacientes es, como se ha señalado, el resultado del no reconocimiento de enfermos con manifestaciones leves, no identificadas o incorrectamente interpretadas.
Como es de esperar, la hipermovilidad articular fue encontrada en una gran cantidad de los familiares afectados y más del 20% tenían historia de luxaciones articulares, muchos de ellos recidivantes.
Las manifestaciones cutáneas se encontraron en la totalidad de los enfermos estudiados. Éstas fueron ligeras y predominó la piel blanda, hiperelástica y aterciopelada, sin evidencia de pseudotumores u otras anomalías dermatológicas.
La hipermovilidad articular estuvo presente en todos los niños estudiados asociada en más de la mitad de ellos a hipotonía muscular evidenciable al examen físico y sintomática en la mayoría. Otros hallazgos articulares como el genus recurvatum, escoliosis y pie plano fueron también encontrados. Es de señalar que un grupo de los pacientes tenían historia de subluxación recidivante, lo que indica el daño articular temprano. La existencia de dolor crónico en los niños, aunque menos frecuente que en los adultos, es una manifestación que requiere suficiente atención para disminuir las limitaciones que con el paso de los años pueden producirse en estos enfermos.
Los prolapsos valvulares no fueron frecuentes y en todos los casos su presencia no evidenció repercusión identificable en la función cardiovascular. El examen oftalmológico mostró la existencia de miopía no acompañada de otras alteraciones oftalmológicas.
Los resultados obtenidos en el coagulograma no mostraron alteraciones dependientes de defectos plasmáticos. El recuento plaquetario fue normal en todos los casos y es de resaltar que tanto la prueba del lazo como el tiempo de sangría, estudios que pueden ser positivos en presencia de alteraciones vasculares o plaquetarias, fueron normales.
El examen de la lámina periférica mostró la presencia de macroplaquetas, plaquetas dispersas y escasa formación de grumos en el 62% de los pacientes estudiados, lo que sugirió la posibilidad de un trastorno cualitativo plaquetario lo que sólo en algunas investigaciones había sido señalado. Los tests de agregación y función plaquetaria presentaron trastornos cualitativos dados por la disminución de la agregación con ADP, sola o combinada con epinefrina y colágeno y con menor frecuencia trastorno de la disponibilidad de fosfolípidos plaquetarios. Es de destacar que la mayor parte de estos pacientes utilizaban de forma continuada antihistamínicos como el ketotifeno por diversas causas, tratamiento que había sido suspendido durante un mes antes de la realización del estudio, lo que indica que el defecto es parte de la enfermedad que probablemente se hace clínicamente relevante con la ingestión del fármaco. De los 181 niños con manifestaciones hemorrágicas a los que se realizó estudio de función y agregación plaquetaria el 69,6% presentó alteraciones de los tests realizados y el 83,4% mostró alteración de la morfología plaquetaria, destacándose su correspondencia con este hallazgo con una elevada frecuencia.25
Formas clínicas
En la primera clasificación del síndrome que se realizó en Berlín en 1986, se describieron 10 subtipos que quedaron posteriormente en desuso por su dificultad para diferenciarlos. En 1997, en Villefranche, se propuso la nueva clasificación, reducida a seis subtipos diferentes según las características clínicas, bioquímicas y genéticas predominantes. Esta clasificación más simplificada con 6 tipos principales está guiada por el principio de que sea útil para los médicos generales. Para cada tipo se han definido criterios diagnósticos mayores y menores. Es necesaria la presencia de uno o más criterios mayores para el diagnóstico clínico o si existe dicho criterio, es altamente indicativo o aconseja confirmación mediante pruebas de laboratorio cuando sea posible. Uno o más criterios menores contribuyen al diagnóstico de un tipo específico de la enfermedad, aunque en ausencia de criterios mayores no serán suficientes para establecer el diagnóstico. La presencia de criterios menores puede ser una sugerencia para el diagnóstico de una entidad parecida al síndrome, la naturaleza de la cual se aclarará cuando las bases moleculares que subyacen en dicha enfermedad se conozcan.
Tipo clásico
Signos mayores: Hiperlaxitud de la piel, cicatrices atróficas extensas, hipermovilidad articular que conduce a los esguinces, luxaciones/subluxaciones, pies planos.
Signos menores: Piel lisa y suave, pseudotumores de tipo moluscos(lesiones carnosas asociadas con cicatrices observadas frecuentemente sobre los sitios de presión), esferoides subcutáneas(pequeños cuerpos duros esféricos subcutáneos a menudo móviles y palpables en los antebrazos y la tibia pueden calcificarse y hacerse visibles radiográficamente), hipoplasia muscular, magulladuras fáciles, extensibilidad y fragilidad tisular que se manifiesta por hernia hiatal, prolapso anal, insuficiencia cervical, hernia postoperatoria, prolapso de la válvula mitral, dilatación de la raíz aortica, rotura prematura de membranas amnióticas.
Este trastorno tiene un patrón de herencia autosómico dominante. 26
El tipo clásico se ha dividido en tipo I (gravis) y tipo II (mitis), que son alélicos. Se consideran idénticos con fenotipos variables.
Representan aproximadamente el 30% de todos los casos. Son pacientes comúnmente prematuros por rotura precoz de las membranas fetales y con frecuentes focos contusivos. Al menos la tercera parte de ellos al nacimiento presentan anomalías congénitas como pie zambo, luxación congénita de cadera o hernia inguinal.
Su diagnóstico suele realizarse al comienzo de la marcha, pues los microtraumatismos reiterados típicos de esta edad pueden producir heridas profundas tanto en los codos como en las rodillas, así como en la cara anterior de la pierna en la vecindad de la cresta tibial anterior. Se pone así de manifiesto la fragilidad de la piel, la cual al ser suturada es cortada por las propias suturas. Esto lleva a unas cicatrices que son prácticamente diagnósticasy deben ser buscadas sobre las eminencias óseas y en general son amplias. Genéticamente se comportan como autosómicas dominantes, desconociéndose cuál es el defecto bioquímico. Entre sus más frecuentes complicaciones figuran la presencia de flebectasias, de roturas viscerales y, en ocasiones, de roturas de vasos mayores.
Prevalencia: Afecta a 1 por 10.000 a 15.000 habitantes.
Tipo hipermóvil
Signos mayores: Piel hiperextensible o piel lisa y suave, hipermovilidad articular generalizada, que afecta más frecuentemente a las articulaciones del hombro, de la rótula y la temporomandibular.
Signos menores: Luxaciones articulares recurrentes, dolor crónico articular de la extremidad, historia familiar positiva, prolapso de la válvula mitral, dilatación de la raíz aórtica.
Tiene un patrón de herencia autosómica dominante.
La piel es suave como terciopelo, laxa, elástica, pálida, con mala cicatrización y a veces con queloides. Otras veces la piel de la cicatriz es delgada como papel (cicatriz papirácea), como la que se ve en cicatrices de vacunas. Pueden presentar telangiectasias, livedo reticularis. Las estrías sin causa aparente pueden estar presentes en personas jóvenes y de preferencia en la zona lumbar. Los pacientes presentan párpados caídos ("cansados"). En ocasiones las articulaciones interfalángicas y los codos presentan piel oscura y arrugada ("codos sucios") y lunares cafés o negruzcos del tamaño de lentejas llamados lunares lenticulares, los que pueden aparecer en cualquier parte del cuerpo, incluso en la cara. Es frecuente encontrar que estos enfermos tienen venas prominentes en el dorso de las manos. Las alteraciones de la piel se ven en la mayoría de los pacientes. La misma puede ser tan típica (mano blanda, suave como terciopelo) que el diagnóstico de síndrome de Ehlers-Danlos tipo III puede sospecharse al darle la mano al enfermo. 27
La fragilidad capilar presente puede originar hematomas recurrentes en presencia de traumatismos leves o sin causa aparente. Ocasionalmente su presencia es de tal magnitud que sugiere maltrato infantil. En los pacientes pediátricos se le suele confundir con la enfermedad de von Willebrand, ya que en ambas los exámenes de coagulación son normales, excepto el tiempo de sangría, que está prolongado en ambos. Se puede diferenciar en que en la enfermedad de von Willebrand existe disminución de dicho factor, lo cual no sucede en el síndrome de Ehlers-Danlos tipio III. Puede haber también historia de epistaxis recurrente o tendencia a sangramiento de las encías.
La hiperlaxitud articular que caracteriza la enfermedad se comprueba en una o más articulaciones asociada a síntomas como dolor, tendinitis subluxaciones entre otros y junto a ello, hiperextensión de los dedos hacia atrás, tocar el antebrazo con el pulgar, el movimiento exagerado de muñecas y codos. Suele existir genu recurvatum.
Lo interesante es que frecuentemente la laxitud afecta sólo una o pocas articulaciones y que la persona no sepa que es hiperlaxa. También es posible padecer la enfermedad sin ser laxo, en caso de tener alteraciones de algunos tejidos (várices, hernias, miopía, etc.) y cumplir con el criterio diagnóstico. 28
A veces la hiperlaxitud es evidente a simple vista en los dedos, codos, muñecas y rodillas. La persona puede tocar el suelo con la palma de las manos o podía hacerlo antes. Cuando niño, divertía a sus amigos contorsionando su cuerpo con posturas extremas, o bien, podía abrirse de piernas en 180°, chuparse el dedo gordo del pie ("niño de goma") o se divertía haciendo "actos malabares" con las manos. Tener alguno de los signos de hiperlaxitud como la posibilidad de extender el dedo meñique a 90°o más; la capacidad de extender los dedos en forma activa adoptando la posición llamada "mano en forma de ave volando" o hacer el "signo del pulgar horizontal", que consiste en colocar el pulgar en posición horizontal en forma activa. Las subluxaciones articulares se producen en la articulación de la base del pulgar, codos, hombros, caderas, rodillas y la articulación temporomandibular, con bruxismo o descarretillamiento. El dolor de espalda puede ser ocasionado por escoliosis, a veces desde la niñez, hiperlordosis por discopatías lumbares o simplemente por laxitud de los ligamentos espinales. Los pacientes son, en ocasiones, altos y delgados (algo gibados), con brazos y pies largos, aracnodactilia, a veces con pectum excavatum o pectum carinatus o con costillas prominentes. Las adolescentes tienen aspecto de modelo, esbeltas, con cuello largo, hombros cuadrados y dedos largos. Este hábito marfanoide es uno de los signos menores del criterio de Beighton para el diagnóstico de este tipo de la enfermedad.
Puede demostrarse densidad mineral ósea baja u osteoporosis en hombres y mujeres jóvenes. Estos enfermos jóvenes con osteoporosis por lo general no sufren fracturas tal como lo hacen las personas con osteoporosis post-menopáusica. 29
Las escleras normales son blancas como el mármol, pero en estos enfermos pueden ser celestes por falta de colágeno se transparenta el plexo coroídeo. Esto ocurre en el 80% de las mujeres, pero en los hombres es menos frecuente y menos notorio. La coloración celeste puede ser muy leve, por lo que hay que buscarla con esmero. Esto no produce ningún problema ocular.
Los pacientes pueden tener historia de neumotórax espontáneo. Esta complicación es más frecuente en el síndrome de Marfán, pero se puede presentar en el en el tipo III de esta enfermedad, así como en el síndrome de Ehlers-Danlos vascular y es más frecuente en hombres que en mujeres. Debido a la debilidad de los tejidos, el pulmón se rompe sin causa aparente o con un traumatismo mínimo.
La artropatía degenerativa es frecuente debido a la presencia de cartílagos débiles y aparece en personas jóvenes, es más destructiva y de evolución rápida.
La presencia de hernias (hiatal, umbilical, inguinal o del núcleo pulposo), várices en pacientes jóvenes, hemorroides, criptorquidia, prolapso vaginal o rectal, prolapso de la válvula mitral, quistes de todo tipo (incluso ganglion de la muñeca y quiste de Baker), constipación, colon irritable e incluso megacolon, son consecuencias de las alteraciones de tejidos blandos.
La lengua puede ser larga, capaz de topar la nariz, muy movible, capaz de hacer un tubo, doblarse de lado y sobre sí misma. La ausencia de frenillo lingual en niños indica que tendrán hiperlaxitud. Estos pacientes pueden presentar acrocianosis no sólo con el frío y calor, sino también relacionada con la inactividad de las manos.
Entre las manifestaciones neurofisiológicas se destacan las alteraciones del sistema nervioso autónomo que pueden producir hipotensión ortostática en la que además participa la alteración del colágeno de la pared venosa de las extremidades, por lo que ésta se dilata contribuyendo a la misma. Es un cuadro muy frecuente, que generalmente no es diagnosticado.
La disautonomía se caracteriza por tendencia a la hipotensión arterial predominantemente ortostática, 30que en ocasiones es compensada por la presencia de taquicardia postural ortostática. Los mareos y pre-síncopes son frecuentes. Son pacientes con intolerancia por el frío, cansancio, fatiga crónica y somnolencia. La diaforesis de las manos es profusa a veces con manos y pies fríos. Puede haber xeroftalmía y xerostomía que requiere diagnóstico diferencial con el Sjögren y colon irritable.
La alteración de la propiocepción es frecuente, lo que debe ser tenido en cuenta durante el tratamiento fisioterapéutico. Los síntomas neuropsiquiátricos incluyen depresión, ansiedad, fobias y crisis de pánico; es frecuente la depresión debido a que los enfermos se sienten mal porque no se les encuentra explicación a sus problemas. También puede existir cefalea, jaquecas, mala memoria, falta de concentración, desorientación, falta de ánimo y calambres.
La facies típica del síndrome de hiperlaxitud articular Se caracteriza por cara triangular con mentón aguzado, escleras celestes, orejas aladas y chicas con hélix chato, sin lóbulo o lóbulo pegado y puntiagudas. La nariz puede ser asimétrica (tabique nasal desviado), con leve prominencia de la unión del cartílago al hueso nasal, con cartílago blando de la punta de la nariz. Los pacientes presentan además párpados caídos u ojos de tipo antimongólico (orientación contraria a los ojos chinos). Es frecuente que los niños presenten retardo motor (dificultad para comenzar a caminar), con hipotonía muscular, a veces son inquietos o francamente hiperactivos, con déficit de atención y concentración, disléxicos, friolentos, con tendencia a hematomas y capaces de hacer "actos malabares" con los dedos, debido a la hiperlaxitud articular. Sus dolores usualmente se atribuyen a "dolores del crecimiento". Generalmente tienen hipotensión, pero a los niños los médicos por lo general no les toman la presión.
Cualquiera de estos síntomas y signos puede comenzar a cualquier edad, incluso durante la niñez. Los niños son más laxos que los adultos y las mujeres más que los hombres. Es importante conocer que tener hiperlaxitud articular sin síntomas es una buena cualidad, pero cuando ésta produce molestias, síntomas y complicaciones se trata de una enfermedad. El síndrome de Ehlers-Danlos tipo III, también denominado síndrome de hiperlaxitud articular es una enfermedad potencialmente seria y no una condición leve. Es necesario que los médicos conozcan que se trata de una enfermedad genética que puede afectar varios tejidos.
Tipo vascular
También es conocido como tipo equimótico del síndrome de Ehlers-Danlos, arterial o tipo Sack-Barabas. Es posiblemente el tipo con mayor incidencia de complicaciones o con complicaciones de mayor gravedad. 31, 32Representa el 4% de todas las formas del síndrome, siendo el de mayor interés en cuanto a las lesiones arteriales, pudiendo ser letal. Su herencia puede ser autosómica recesiva o dominante.
Signos mayores: Piel delgada y traslúcida, fragilidad o rotura arterial/intestinal/uterina, magulladuras extensas, apariencia facial característica que incluye una nariz delgada y fina, labios delgados, piel tensa, mejillas hundidas y ojos prominentes secundaros a déficit de tejido adiposo.
Signos menores: Hipermovilidad de las articulaciones pequeñas, rotura de tendón y musculo, rotura de vejiga urinaria, pie torcido equino varo, venas varicosas, fistulas arteriovenosas y del seno carotídeo y cavernoso, neumotórax/hemotórax, recesión gingival, historia familiar positiva.
Tiene un patrón de herencia autosómica dominante. Son responsables las mutaciones en el gen COL3A1 que conducen a defectos en la cadena procx (III) del colágeno tipo III.
El cuadro clínico se caracteriza por una gran tendencia a las equimosis y focos contusivos, expresión de la tendencia a la ruptura espontánea de grandes vasos y de vasos de mediano tamaño, así como a rupturas espontáneas de intestino. La frecuencia de las complicaciones vasculares parece obedecer a que los pacientes de este grupo pueden sintetizar cantidades variables de colágeno tipo III, que en la pared vascular es el responsable de su integridad estructural y de su resistencia a la tensión, siendo a la vez un elemento de interacción colágeno-plaqueta y en la formación del trombo. Su prevalencia es de 1/100, 000.
De las 4 variedades de colágena existentes en el cuerpo, el tipo III se encuentra en los vasos, piel, pulmón e intestino. Clínicamente se caracteriza por presentar una hipermovilidad articular limitada a los dedos, y el mayor grado en las articulaciones interfalángicas distales de los dedos. La hipermovilidad de las otras articulaciones es poco llamativa. La piel no es excesivamente extensible, pero sí es muy fina, expresando un déficit de colágena en el corion, que provoca que el sistema venoso subcutáneo se haga muy visible, lo cual sucede particularmente en el tronco. La piel de la cara, orejas y de los dedos suele estar tirante. Muchos pacientes sufren rupturas de la piel.
Tipo artrocalasia
Signos mayores: Hipermovilidad articular generalizada grave con subluxaciones mrecurrentes, luxación congénita de cadera.
Signos menores: Hiperlaxitud de la piel, fragilidad tisular, magulladuras fáciles, hipotonía muscular, cifoscoliosis, osteopenia.
Tiene un patrón de herencia autosómica dominante. Son responsables las mutaciones en los genes COL1A1 y COL1A2, que codifican las cadenas α1 y α2 del colágeno tipo I.
Tipo dermatoparaxis
Signos mayores: Fragilidad cutánea importante, piel redundante hundida.
Signos menores: Piel blanda macerada, magulladuras fáciles, rotura prematura de membranas amnióticas, hernias umbilicales e inguinales.
Tiene un patrón de herencia autosómico recesiva. Son responsables las mutaciones homocigóticas o componentes heterocigóticos en el gen que codifica la peptidasa N- terminal del procolágeno tipo I. Resulta diagnostica la determinación electroforética de las cadenas pNα1 (I) y pNα2 (I) del colágeno tipo I extraído de la dermis en presencia de inhibidores de la proteasa u obtenido de los fibroblastos.
Tipo cifoescoliosis
(La presencia de 3 o más criterios mayores en la infancia es diagnóstica).
Signos mayores: Laxitud articular generalizada, hipotonía muscular intensa desde el nacimiento, escoliosis progresiva con inicio en el nacimiento, fragilidad escleral y rotura del globo ocular.
Signos menores: Fragilidad tisular, magulladuras fáciles, rotura arterial, hábito marfanoide, microcórnea y osteopenia.
Tiene un patrón de herencia autosómico recesiva. La deficiencia de lisil-hidroxilasa (PLOD), una enzima que modifica el colágeno, es la responsable.
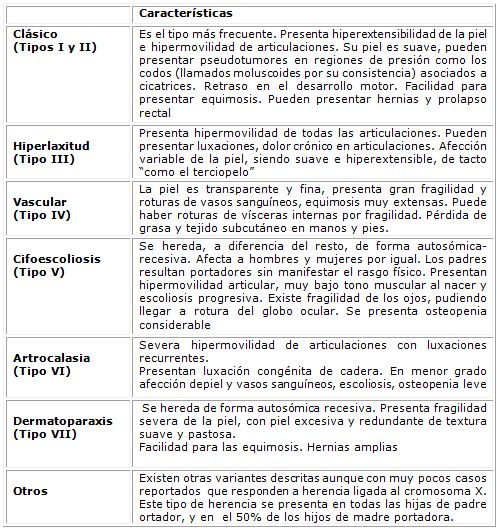
En la Tabla 1 se resumen las características clínicas de los diferentes tipos de síndrome de Ehlers-Danlos.
Tabla 1: Tipos de Elhers Danlos y sus características clínicas.
Diagnóstico positivo
El diagnóstico del síndrome de Ehlers-Danlos es limitado ya que en la actualidad no hay ninguna prueba sencilla, objetiva y fiable que permita diagnosticar todos y cada uno de sus tipos. Muchas personas con la enfermedad requieren un diagnóstico clínico confirmado habitualmente por un genetista especializado en enfermedades raras. Especialistas como dermatólogos, pediatras y reumatólogos también pueden diagnosticar el trastorno.
Para diagnosticarlo correctamente los profesionales deben valorar los antecedentes familiares y la historia clínica del enfermo. Como fue señalado, la mayor parte de los enfermos tienen una mezcla de síntomas de todas las categorías y no entran en una categoría claramente definida. La severidad de las manifestaciones clínicas puede variar incluso entre los miembros de la misma familia, e incluso cada persona de la misma familia puede estar afectada de manera diferente.
Debido a esta dificultad para el diagnóstico, al hecho de que se considera una enfermedad rara y a que la hiperlaxitud articular es un signo clínico que muchas veces por obvio, los profesionales de la salud no tienen en cuentaque el síndrome de Ehlers-Danlos es una enfermedad que a menudo pasa desapercibida siendo en la actualidad subdiagnosticada.
Criterios de hiperlaxitud articular
La hiperlaxitud articular se valora según diversas escalas. Se calcula que aproximadamente un 10% de la población tiene hiperlaxitud articular, es decir, son capaces de mover una o más de sus articulaciones de forma exagerada. Aunque la mayoría de estas personas hiperlaxas no tienen ningún síntoma, ni sufren ninguna enfermedad definida. A pesar de ello, los expertos opinan que cualquier articulación hiperlaxa puede sufrir las consecuencias de este trastorno independientemente de cuál sea su causa. Por otro lado, la hiperlaxitud articular es también un signo clínico que forma parte de otras enfermedades como el síndrome de Marfan, la osteogénesis imperfecta y el síndrome de Down.
Beighton y colaboradores 6establecieron estos criterios, proporcionando una escala de evaluación que es la más utilizada hoy en día y es considerada aún el pilar fundamental de los criterios que se han propuesto hasta ahora. La escala de Beighton otorga al paciente 1 punto por cada una de las siguientes características:
Extensión pasiva de la quinta metacarpofalange que sobrepase los 90 grados.
Aposición pasiva del pulgar al antebrazo.
Hiperextensión del codo de más de 10 grados.
Hiperextensión de la rodilla de más de 10 grados.
Flexión del tronco que permita que las palmas de la mano se posen en el suelo.
En dichos criterios se puntualiza que cada extremidad de los cuatro primeros es un item por separado según fuera la derecha o la izquierda, generando una posible puntuación de 9.
No hay un acuerdo general, ni universal sobre el mínimo de puntuación para el diagnóstico de hiperlaxitud articular; algunos investigadores usan la puntuación de Beighton de 6/9,61e incluso otros usan una puntuación modificada de Beighton de 3/5.
Criterios de hiperextensibilidad de la piel
El grado de hiperextensibilidad de la piel varía de una persona afectada a otra, e incluso puede variar de una parte del cuerpo a otra. El punto más adecuado para medir la hiperextensibilidad de la piel es generalmente la parte ventral del antebrazo en el punto medio entre el codo y la muñeca. La distancia a la cual este pliegue se puede extender sin causar molestias se mide en centímetros.
Se calcula una puntuación de 0 a 5 puntos sobre las bases siguientes:
0 = Menos de 4cm 0: ≤ de 4 cm
1 = Más de 4 cm 1: ≥ de 4 cm
2 = 5 cm 2: 5 cm
3 = 6 cm 3: 6 cm
4 = 7 cm 4: 7 cm
5 = Más de 8 cm 5: ≥ de 8 cm
La hiperextensibilidad de la piel debe examinarse en un área neutral, es decir, en un punto en el que la piel no esté sometida a fuerzas mecánicas o tenga cicatrices, por ejemplo, en la superficie volar del antebrazo. Se mide estirando la piel hasta que se perciba resistencia. En niños pequeños puede ser difícil de medir debido a la abundancia de tejido graso subcutáneo. La hiperlaxitud articular depende de la edad, el sexo, la familia y los antecedentes étnicos. Una puntuación de 5/9 o más define hiperlaxitud.
La facilidad para las magulladuras manifestada por equimosis espontáneas, frecuentemente recurrentes en las mismas áreas, causan la característica decoloración pardusca de la piel. La facilidad para las lesiones puede ser el síntoma de presentación del síndrome de Ehlers-Danlos en la temprana infancia. La fragilidad de los tejidos se manifiesta como facilidad para las equímosis y la presencia de cicatrices atróficas. Las cicatrices se encuentran principalmente en los puntos de presión (rodillas, codos, frente) y tienen una apariencia papirácea y atrófica. Con frecuencia estas cicatrices se vuelven amplias y descoloridas; la cicatrización suele estar comprometida y es muy lenta y tórpida. El prolapso de la válvula mitral y la dilatación de la aorta proximal deben diagnosticarse mediante un ecocardiograma. Es una manifestación muy frecuente, pero la dilatación de la aorta no es tan habitual. Deben utilizarse criterios exigentes para el diagnóstico del prolapso de la válvula mitral.
Aunque bien definidos, los tipos artrocalasia, cifoescoliosis y dermatoparaxis son considerablemente menos frecuentes que los tipos clásico, hiperlaxitud y vascular.
Diagnóstico diferencial
La hiperlaxitud articular es una manifestación no especifica de amplio grupo de diferentes enfermedades y síndromes, entre los cuales el síndrome de Elhers Danlos puede distinguirse por las características halladas y por la inclusión de otros aparatos diferentes de la piel y las articulaciones. Algunos ejemplos incluyen:
• Aneupliodes, tales como los síndromes de Down y de Turner.
• El síndrome de Williams (síndrome de deleción en el gen que codifica la elastina) y la estenosis aortica supravalvular causada por una mutación en el gen que codifica la elastina.
• Enfermedades del colágeno y otras displasias esqueléticas tales como el síndrome de stickler, la osteogénesis imperfecta, la acondroplastia y la hipoacondrolastia.
• El síndrome del Marfan y las enfermedades relacionadas
Complicaciones
Las complicaciones al igual que las manifestaciones clínicas habituales del síndrome son debidas a la existencia de un aumento de la elasticidad de los tejidos en el organismo, lo que los hace incompetentes o frágiles. Entre las distintas complicaciones se observan:
Neurológicas
Son raras. Se han descrito neuropatías del plexo braquial y lumbosacro por presiones anómalas propiciadas por la laxitud ligamentosa. En otras ocasiones la laxitud, por el contrario, facilita la lesión neural, como el caso comunicado por El-Shaker, en el que la secuela fue una neuropatía aguda del plexo braquial al utilizar tracción mediante halo después de una liberación anterior corrigiendo una escoliosis.
El tipo vascular se asocia a accidentes vasculares encefálicos por rupturas vasculares que son más frecuentes después de la adolescencia.
Intestinales
a) Ectasia colónica
b) Rotura del colon sigmoides.
c) Rotura del recto.
d) Hemorragias digestivas
e) Hernias
f) Divertículos intestinales
Cardíacas
Las anormalidades cardíacas descritas en el síndrome de Ehlers-Danlos son asociaciones fortuitas, casuales o muy infrecuentes.
Vasculares
Están presentes en el síndrome de Ehlers-Danlos tipo I o "gravis" y en el tipo IV o "arterial" Dentro de ellas podemos encontrar:
a) Roturas arteriales mayores espontáneas o ante mínimos traumatismos.
b) Formación de aneurismas intracraneales, en la aorta o arterias periféricas.
c) Fístulas arteriovenosas carotidocavernosas o aortocava.
Debido a las características propias del síndrome se deben tener en cuenta especiales consideraciones técnicas para tratar las posibles complicaciones vasculares. Como consecuencia de la fragilidad de los vasos sanguíneos no es posible suturarlos, por ello se recomienda la compresión externa del punto de hemorragia cuando la hemorragia se localiza en un miembro, y en otros sitios, como la subclavia u otros vasos que no pueden ser comprimidos, se aconseja la ligadura con cinta umbilical y clips metálicos. El control proximal del vaso se debe realizar con torniquetes porque los clips vasculares pueden producir roturas arteriales.
La anticoagulación puede producir grandes hematomas, sobre todo en el tipo IV, por lo que se debe evitar.
Cutáneas
Las heridas en el síndrome de Ehlers-Danlos tienen una curación lenta originando cicatrización asimétrica, lo que produce cicatrices anchas, atróficas, delgadas, brillantes y con frecuencia hiperpigmentadas y surcadas por arrugas. La escisión de la cicatriz y su resutura o injerto cutáneo ha demostrado ser un tratamiento eficaz.
Dehiscencia de la herida. Es debida a la lenta curación de las heridas en el síndrome y a la dificultad de sutura de la piel como consecuencia de su fragilidad. A veces se presenta de forma recurrente.
Siempre ante toda cirugía se debe examinar la piel del paciente para detectar variaciones en su textura y poder de esta forma tomar las precauciones necesarias y explicar al paciente la posibilidad de presentar cicatrices antiestéticas. El cierre de la herida debe ser siempre meticuloso, y si la sutura rompe los bordes de la piel se pueden utilizar en su lugar bandas adhesivas o bien puntos de gran retención.
Ulceraciones cutáneas. Pueden aparecer de forma espontánea sobre la espina tibial o el codo. A veces requieren injertos cutáneos para su curación.
Tratamiento
No hay tratamiento curativo del síndrome de Elhers Danlos, por lo que el tratamiento es sintomático. El ingerir colágeno no ayuda. Es posible que en el futuro la terapia génica, sea una solución.
Tratamiento general
Medidas preventivas
Puede conseguirse mejorar la estabilidad mediante ejercicios de baja resistencia para aumentar el tono muscular (contracciones musculares en reposo, contrapuertas a las voluntarias). Ejemplos de este tipo de ejercicios incluye andar, hacer bicicleta, ejercicios aeróbicos de bajo impacto, nadar o hacer ejercicios en el agua y ejercicios de amplitud de movimiento sin resistencia añadida. El progreso debe hacerse mediante el aumento de repeticiones, su frecuencia o duración y no mediante incremento de la resistencia. Puede costar meses o años que se pueda reconocer un progreso significativo.
La hiperextensión articular debe evitarse las personas con hiperlaxitud necesitan generalmente que las eduquen sobre el rango normal del movimiento y que les enseñen a tener precaución en no sobrepasarlo. Los ejercicios de alta resistencia y actividades de alto impacto aumentan el riesgo de subluxaciones y dislocaciones articulares agudas, el dolor crónico y la osteoartritis. Por lo tanto, los deportes de alto impacto como el futbol están contraindicados. De todas maneras, la mayoría de los deportes y actividades son aceptables si se toman las precauciones adecuadas.
Utilizar utensilios gruesos, para escribir puede reducir el estrés en los dedos y en las articulaciones de las manos, por ejemplo, un asa no convencional en el utensilio de escribir puede dar como resultado una disminución sustancial del estrés axial en las articulaciones interfalángicas, metacarpofalángicas y carpometacarpianas. Estos ajustes con frecuencia dan como resultado una reducción marcada del dolor en el dedo índice y en la base del pulgar.
Tratamiento específico
Tratamiento de la fase aguda
Es necesario el reposo de la articulación afectada. Por lo general se requiere aplicación de calor o frío y a veces el uso de férulas. Son útiles los masajes, ultrasonido y ultratermia. Es necesario evitar la actividad repetitiva que produjo una lesión. Se recomienda paracetamol o antiinflamatorios por períodos cortos. Los relajantes musculares son de ayuda y las infiltraciones con esteroides están indicadas en las bursitis y tendinitis.
Tratamiento de la fase crónica
El reumatólogo es el especialista más indicado para tratar estos pacientes por su conocimiento, ya que es necesario ser capaz de diferenciar el síndrome de hiperlaxitud articular de las otras formas más graves como el síndrome de Marfan y la osteogénesis imperfecta. A pesar de que el dolor es el principal síntoma del mismo, los antiinflamatorios son de efecto limitado y es preferible usar analgésicos.
Kinesiterapia: La terapia ocupacional es muy útil en caso de ser efectuada por un terapeuta con conocimientos sobre la hipermovilidad articular, de lo contrario los resultados no son buenos. El principal logro es restablecer el rango de movilidad normal de la articulación, corregir la disfunción del movimiento, mejorando la estabilidad articular y la condición física general. Los ejercicios deben fortalecer los músculos y tendones. Ocasionalmente es necesario inmovilizar alguna articulación, pero solamente durante el episodio agudo. La acupuntura ocasionalmente puede ser necesaria como tratamiento en una clínica de control del dolor.
Medicamentos: Se recomienda a estos pacientes tomar de 0,4 a 1 mg de ácido fólico diarios en forma permanente, ya que hay estudios que demuestran que la deficiencia de folato altera los enlaces del colágeno, produciendo debilidad de los tejidos. Para el manejo del calambre es útil el magnesio y en caso de fragilidad capilar, además de evitar contusiones, es necesario indicar vitamina C o un polivitamínico. Es importante detectar y tratar precozmente la osteoartritis. El uso de glucosamina sola o con condroitin sulfato, para prevenir la artrosis, es sugerido por algunos y parece ser útil, pero requiere de estudios que lo confirmen. El tratamiento de la osteoartritis debe iniciarse lo antes posible de la forma usual.
Tratamiento de la disautonomía: El problema disautonómico principal es la hipotensión postural que puede ser tratada con medidas generales como ingerir 2 a 3 litros de líquidos al día, aumentar la ingesta de sal (excepto contraindicaciones), usar medias elásticas hasta la rodilla y reposar después de almuerzo. Se debe caminar despacio evitando la inactividad y la bipidestación prolongada. De existir anemia o deshidratación, es necesario corregirlas. Estas medidas generales asociadas a medicamentos para elevar la presión arterial, tales como la etilefrina, midodrina y la fluodrocortisona, pueden mejorar significativamente la calidad de vida del paciente. 35
Tratamiento en equipo: En ocasiones es necesaria la opinión de un cardiólogo, ginecólogo u otro especialista, incluyendo psicólogos, psiquiatras, fisiatras, kinesiólogos y terapeutas ocupacionales. Los grupos multidisciplinarios son de ayuda considerable, tanto para los enfermos como para los familiares. Ocasionalmente se requiere cirugía, como en el caso de subluxaciones recurrentes, pero es necesario recordar la mayor tendencia de hemorragias, infecciones y mala cicatrización en estos enfermos.
Tratamiento del dolor
La medicación del dolor debe monitorizarse según los síntomas subjetivos de la persona afectada y no por los hallazgos objetivos. Muchos médicos aconsejan un especialista en el tratamiento del dolor, pero el dolor podría tratarlo el médico de cabecera utilizando alguno de los siguientes fármacos:
• Acetaminofeno (paracetamol) 10mg/kg/día en 3 o 4 dosis diaria puede ser útil y bien tolerado.
• Fármacos antiinflamatorios no esteroideos (AINES), por ejemplo, ibuprofeno. Deben valorarse hasta la dosis máxima o hasta la dosis bien tolerada según los síntomas del sistema gastrointestinal.
• Se puede añadir tramadol al paracetamol más un AINE antes de recurrir a otros opiodes. Las náuseas son el efecto secundario más frecuente.
• La lidocaína tópica en crema o parches puede ser útil, a veces en zonas con calor localizado.la capsaicina tópica tiene una utilidad cuestionable, pero es segura.
• Los relajantes musculares esqueléticos son útiles, en combinación con los fármacos anteriores citados, para tratar los espasmos miofasciales. La metaxalona (Skelatin), puede producir menos sedación, pero todos los relajantes pueden producir algún grado de sedación.
• Dosis bajas de antidepresivos tricíclicos a menudo son efectivas para dolor neuropático con beneficios adicionales de sedación leve y puede mejorar un poco el humor. Las dosis típicas son nortriptilina (25-100 mgs) y la trazadona (20-25 mgs) cada noche.
• Algunas medicaciones anticonvulsivas son también efectivas para el dolor neuropático y puede usarse conjuntamente con antidepresivos tricíclicos; todos requieren un aumento gradual antes de alcanzar niveles terapéuticos. La gabapeptina se comienza habitualmente con 300 mgs 3 veces al día y debería ir aumentándose progresivamente antes de decidir que no es útil. El topiramato y la lamotrigina también pueden ser útiles.
• Los opioides son efectivos tanto para el dolor miofacial como para el dolor neuropático, pero normalmente se reservan tanto como sea posible. Pueden administrarse al mismo tiempo que todos los anteriores excepto el tramadol. Puesto que generalmente se usan de forma crónica, la formulación primaria debe hacerse con un fármaco de larga acción combinado con fórmulas de corta acción usadas solamente para romper el ciclo del dolor. El uso rutinario de 2 o más dosis diarias de un fármaco de acción corta debería apuntar a un aumento en la dosis de un fármaco de acción larga u otro ajuste para el tratamiento del dolor.
Manejo de los trastornos hemostáticos
En los pacientes con trastornos hemostáticos o tendencia al sangramiento debe tenerse en cuenta que las equimosis fáciles o espontaneas, generalmente no requieren del mismo. La existencia de un tiempo prolongado de sangrado puede aconsejar una evaluación para la enfermedad de Von Willebrand, que generalmente es negativa. En caso de sangrado severo o profilaxis quirúrgica, puede resultar útil el acetato de desmopresina.
En los niños con síndrome de Ehlers-Danlos la presencia de trastornos de la agregación y función de las plaquetas tiene relevancia clínica variable y la tendencia a presentar manifestaciones hemorrágicas espontáneas puede ser potencializada por el empleo de drogas que alteren estas funciones, entre ellas los antihistamínicos, que se utilizan frecuentemente en la edad pediátrica, en particular ketotifeno, que son drogas que se sabe inhiben reversiblemente la síntesis de prostaglandinas por lo que se recomienda que el uso de estos fármacos, muchas veces indiscriminado, debe ser cuidadosamente evaluado debido al riesgo potencial que ello implica a causa de los defectos hemostáticos señalados.
Al igual que sucede en todo paciente con trastornos cualitativos plaquetarios, el empleo de drogas anti-inflamatorias u otras que alteren su función deben ser utilizadas con precaución.
El empleo de hemoderivados en estos enfermos debe reservarse para aquellos que presentan manifestaciones clínicas con sangrado espontáneo cutáneo-mucoso o visceral, lo cual es excepcional. En aquellos con tendencia hemorrágica leve o moderada, acompañada de anomalías de la función o agregación plaquetaria, el uso de drogas antifibrinolíticas como el ácido tranexámico de forma profiláctica ha demostrado ser suficiente para evitar hemorragias durante la realización de procederes invasivos, todo lo cual indica que la evaluación clínica individual es el criterio que define el manejo de los trastornos hemostáticos en estos pacientes, mientras que el empleo de hemoderivados debe ser definido por el hematólogo debido a los riesgos que implica la terapia transfusional.
Otras acciones terapéuticas
-
La gastritis o los síntomas de reflujo pueden requerir tratamiento. Se deben investigar otras causas tratables como la infección por helicobacter pylori. Debe identificarse el retraso en el vaciado gástrico y si está presente, tratarse con agentes procinéticos como la metoclopramida.
-
El síndrome de intestino irritable se trata usualmente con antiespasmódicos, antidiarreicos y laxantes según se necesite. En casos excepcionales puede ser necesaria una colonoscopia para descartar otros diagnósticos tratables.
-
En estos pacientes es útil realizar un ecocardiograma basal para evaluar el diámetro de la raíz aórtica ajustado según edad y superficie corporal. El pronóstico a largo plazo y el intervalo para la repetición de esta evaluación es por el momento desconocida. Los betabloqueantes pueden valorarse en caso de progresión de dilatación de la raíz aortica. Una dilatación severa requerirá intervención quirúrgica.
-
La hipotensión mediada neuralmente y la taquicardia postural ortostática se tratan como habitualmente con sodio y agua para expandir el volumen de sangre, betabloqueadores, fludrocortisona y/o estimulantes.
-
Los aparatos ortodónticos y las correcciones del paladar suelen tender a caerse. Se debe identificar y tratar la enfermedad peridontal. La laxitud de la articulación temporomandibular y su disfunción son fáciles de tratar. El descanso oral (minimizando la masticación y el hablar) la liberación miofacial y los relajantes musculares pueden ser beneficiosos durante crisis agudas.37
-
Finalmente, la evaluación de los síntomas psiquiátricos en las personas afectadas puede ser de una ayuda inmensa ya que muchos de ellos han sido causados por médicos que durante la atención previa han subestimado malinterpretado o ignorado sus manifestaciones clínicas, lo que origina trastornos como ansiedad, depresión y otros. 36
-
La depresión es un resultado frecuente del dolor crónico y otras complicaciones. El consejo psicológico y orientado al dolor puede mejorar la adaptación y la aceptación a estos problemas y a las necesarias limitaciones físicas. Los antidepresivos también pueden ser beneficiosos. 74
-
La vitamina C es un cofactor en el metabolismo de las fibrillas de colágeno. Se recomienda el uso de un suplemento de 500 mgs/día y/o el consumo de frutas frescas para mejorar algunas de las manifestaciones, así como la utilización de multivitaminas y suplementos naturales como la miel de abejas.
Consideraciones finales
La mayoría de los pacientes con síndrome de Ehlers-Danlos suelen manifestar características clínicas similares (laxitud articular o elasticidad cutánea), pero no todos cumplen con los criterios principales de los subtipos; por lo tanto, se encuentra subdiagnosticado. Se requieren estudios genéticos y bioquímicos que ayuden a identificar los defectos coexistentes. Algunas formas del síndrome son comunes, mientras que otras muy raras. Es importante realizar la exploración física completa y buscar las características específicas de cada subtipo para integrarlo lo más preciso posible. Las pacientes con subtipo vascular deben recibir asesoría, ya que tienen mayor riesgo de padecer complicaciones fatales. El síndrome de Ehlers-Danlos puede ser un potencial debilitador. Los pacientes requieren medidas preventivas y protectoras desde el nacimiento. Es importante asesorar a los familiares para que conozcan las complicaciones asociadas, a fin de preservar la función articular, minimizar el impacto físico y emocional y mejorar su calidad de vida.
Es necesario que padres, profesores, entrenadores, terapeutas físicos, médicos y psicólogos conozcan la alta frecuencia y complicaciones asociadas a la hipermovilidad articular. También es importante informar a la opinión pública, a fin de aumentar la alerta diagnóstica de esta condición poco conocida, que parece ser la causa de las lesiones recurrentes de niños.
Pocos médicos tienen presente que la hiperlaxitud no tiene porqué aparecer en todas las articulaciones de la persona afectada. Algunos especialistas opinan que incluso si una sola articulación hiperlaxa causa dolor o inestabilidad el diagnóstico debe seguir siendo síndrome de hiperlaxitud articular.
Otro error común es considerar que los síntomas aparecen en la infancia, lo cierto es que son muchos los afectados que han manifestado sus síntomas en la adolescencia y/o en la edad adulta, pudiendo llevar hasta entonces una vida normal.
Este desconocimiento de la enfermedad es común y suele provocar un retraso, generalmente de años, en el diagnóstico correcto, así como la aplicación de tratamientos inadecuados. Todo ello, unido a otros factores inherentes a esta patología (el dolor, las discapacidades, etc.) ha provocado en no pocos afectados frustración, ansiedad y depresión. Como consecuencia, a menudo la depresión y/o ansiedad se ven como la causa de la enfermedad, cuando en realidad son su resultado.
Debe lograrse una sensibilización, no sólo de la opinión pública, sino también de las autoridades y organismos competentes, sobre las múltiples discapacidades y manifestaciones que estos síndromes pueden generar.
Saber es comprender, y aunque todavía queda un largo camino por recorrer, comprender estas condiciones es avanzar, y el conocimiento ganado puede ayudar a las personas afectadas a convivir con su enfermedad y en definitiva a tener una mejor calidad de vida.
Desde el año 2011 se aplica en Pinar del Río una guía de acción terapéutica para mejorar el diagnóstico y manejo terapéutico de esta entidad en la población pediátrica, lo que ha permitido establecer acciones de salud por los médicos de la atención primaria y las especialidades afines según la expresión clínica variable de estos enfermos.
Las investigaciones realizadas en el contexto de un proyecto ramal de investigación evidenciaron por primera vez la existencia de un trastorno cualitativo plaquetario como expresión de la enfermedad y su relación con la utilización de terapia anti-histamínica en niños, lo cual ha permitido tomar medidas para disminuir la tendencia hemorrágica en estos pacientes. El manejo multidisciplinario de los mismos ha permitido realizar acciones de salud encaminadas a mejorar la calidad de vida de los enfermos.
Es necesario que padres, profesores, entrenadores, terapeutas físicos, médicos y psicólogos conozcan la alta frecuencia con que dichos pacientes presentan hipermovilidad articular y las complicaciones que de ella se derivan. Las charlas educativas y otras actividades de educación para la salud, pueden ser útiles para informar adecuadamente a la población a fin de aumentar sus conocimientos acerca de esta enfermedad poco conocida.
REFERENCIAS BIBLIOGRÁFICAS
1. Parapia L A, Jackson C. Ehlers-Danlos syndrome. A historical review. Br J Haematol [internet] 2008[citado 2008 apr]; 141(1):[Aprox.3p].Disponible en : https://www.ncbi.nlm.nih.gov/pubmed/18324963
2. Tschemogubow AN. Ein fall von cutis laxae. Protokoly Moskowskawo Venereoologits cheskawo Dermatlogits cheskawo. Obtschestwa ;1891.
3. Danlos M. Un caso de cutis laxa avectumeurs par contusioncronique des coudes et des genoux (xanthomejuvenilepseudo-diabetique de MM. Hallopeau et Marce deLepinay). Bull SocFrançaisDermatolSyphilig; 1908.
4. Campo Díaz M.C, Fortún Campo A., Beades Martínez A., Gato Santiesteban Y., Valdés Sojo C. Caracterización del síndrome de Ehlers-Danlos tipo III. Rev Ciencias Médicas [Internet]. 2013 Jun [citado 2017 jun 20]; 17(3): [Aprox.8p]. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1561-31942013000300003&lng=es
5. Alfaro K. , Martínez M. ,Chirinos F., Muñoz R. "Los Síndromes de Ehlers-Danlos según la clasificación de Villefranche (1997)" EHLERS DANLOS SYNDROMES: Clasificación Revisada , Villefranche , 1997. ODOUS científica.[intenet] 2013[citado 2013 dic 19];171(1).Disponible en: https://www.researchgate.net/publication/237509735_Los_Sindromes_de_Ehlers-Danlos_segun_la_clasificacion_de_Villefranche_1997_EHLERS-DANLOS_SYNDROMES_Clasificacion_Revisada_Villefranche_1997
6. Beighton P, Grahame R, Bird HA. Hypermobility of Joints. 4th edition. London, UK: Springer; 2012.
7. Klemp P, Williams SM, Stansfield SA. Articular mobility in Maori and European New Zealanders. Rheumatology.[internet] 2002 [citado 2002 may];41(5):[Aprox.3p].Disponible en : https://www.ncbi.nlm.nih.gov/pubmed/12011380
8. Castori M, Morlino S, Celletti C, Ghibellini G, Bruschini M, Grammatico P, et al. Re-writing the natural history of pain and related symptoms in the joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type. Am J MedGenet A. [internet] 2013 [citado 2013 dic]; 161ª(12):[Aprox.15p].Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/24254847
9. Voermans NC, Knoop H. Both pain and fatigue are important possible determinants of disability in patients with the Ehlers-Danlos syndrome hypermobility type. Disability and Rehabilitation.[internet] 2011[citado 2010 nov 15];33(8):[Aprox.1p]. Disponible en : https://www.ncbi.nlm.nih.gov/pubmed/21077749
10. Castori M, Morlino S, Celletti C, Celli M, Morrone A, Colombi M, et al. Management of pain and fatigue in the joint hypermobility syndrome (a.k.a. Ehlers-Danlos syndrome, hypermobility type): principles and proposal for a multidisciplinary approach. Am J MedGenet A. [internet] 2011[citado 2010 nov 15]; 158ª(8):[Aprox.15p].Disponible en : https://www.ncbi.nlm.nih.gov/pubmed/21077749
11. Quispe Pari G D. Sindrome de EhlersDanlos (SED). Rev. Act. Clin. Med [Internet]. 2014 [citado 2015 Sep 21]; 45. Disponible en: http://www.revistasbolivianas.org.bo/scielo.php?script=sci_arttext&pid=S2304-37682014000600002&lng=es
12. Campo Díaz M C, Hernández González J L, Gato Santiesteban Y, Valdés Sojo C, Fortún Prieto A. Evaluación de la hemostasia en niños con Síndrome de Ehlers-Danlos tipo III. Rev Cubana Hematol Inmunol Hemoter [Internet]. 2014 Jun [citado 2015 Sep 21] ; 30(2):[Aprox. 8p]. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892014000200007&lng=es
13. Bravo JF, Wolff C. Clinical study of hereditary disorders of connective tissues in a chilean population: joint hypermobility syndrome and vascular Ehlers-Danlos syndrome. Arthritis and Rheumatism[internet]2006[citado 2006 feb] ;54(2):[Aprox.8p].Disponible en : https://www.ncbi.nlm.nih.gov/pubmed/16447226
14. Castori M, Morlino S, Ritelli M, Brancati F, de Bernardo C, Colombi M, et al. Late diagnosis of lateral meningocele syndrome in a 55-year-old woman with symptoms of joint instability and chronic musculoskeletal pain. Am J MedGenet A.[internet] 2014[citado 2013 dic 05];164A (2):[Aprox.6p].Disponible en : https://www.ncbi.nlm.nih.gov/pubmed/24311540
15. Baumann M, Giunta C, Krabichler B, et al. Mutations in FKBP14 cause a variant of Ehlers-Danlos syndrome with progressive kyphoscoliosis, myopathy, and hearing loss. The American Journal of Human Genetics.[internet] 2012[citado 2012 feb 10] ; 90(2):[Aprox.5p]. Disponible en : https://www.ncbi.nlm.nih.gov/pubmed/22265013
16. De Paepe A, Malfait F. The Ehlers-Danlos syndrome, a disorder with many faces. Clinical Genetics.[internet] 2012[citado 2012 jul ]; 82(1):[Aprox. 10p].Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/22353005
17. Bravo Jaime F. Síndrome de Ehlers-Danlos con especial énfasis en el síndrome de hiperlaxitud articular. Rev. méd. Chile [Internet]. 2009 Nov [citado 2015 Sep 21] ; 137( 11 ):[Aprox.9p]. Disponible en: http://www.scielo.cl/scielo.php?script=sci_arttext&pid=S0034-98872009001100013&lng=es. http://dx.doi.org/10.4067/S0034-98872009001100013
18. Easton V, Bale P, Bacon H, Jerman E, Armon K, Macgregor AJ. A89: the relationship between benign joint hypermobility syndrome and developmental coordination disorders in children. Arthritis Rheumatol [internet] 2014[citado 2014 mar ] ;66(Suppl 11): S124. Disponible en : https://geneticamente-incorrecta.blogspot.com/p/manifestaciones-neurologicas-en-el.htm l
19. Castori M, Sperduti I, Celletti C, Camerota F, Grammatico P. Symptom and joint mobility progression in the joint hypermobility syndrome (Ehlers-Danlos syndrome, hypermobility type) ClinExpRheumatol [internet]2011 [citado 2011 nov- dic ] ;29(6):[Aprox.7].Disponible en : https://www.ncbi.nlm.nih.gov/pubmed/22208933
20. Hayward C. Clinical approach to the patient with bleeding or bruising. En: Hoffman: Hematology, basic principles and practice. 6th ed. Philadelphia: Churcill Livingstone Elsevier;2013 . 1847-56.
21. Gharbiya M, Moramarco A, Castori M, et al. Ocular features in joint hypermobility Syndrome/Ehlers-Danlos syndrome hypermobility type: a clinical and in vivo Confocal Microscopy Study. American Journal of Ophthalmology [internet] 2012[citado 2012 sep ] ;154(3):[Aprox. 7p].Disponible en : https://www.ncbi.nlm.nih.gov/pubmed/22633352
22. Maricq HR, Carpentier PH, Weinrich MC, Keil JE, Franco A, Drouet P et al. Geographic variation in the presence of Raynaud's phenomenon. Charleston, SC, USA vs. Tarentaise, Savoie, France. JRheumatol [internet]1993[citado 1993 feb]; 20(1):[Aprox.6p].Disponible en : https://www.researchgate.net/publication/14758480_Geographic_variation_in_the_prevalence_of_Raynaud%27s_phenomenon_Charleston_SC_USA_vs_Tarentaise_Savoie_France
23. Lammers K, Lince SL, Spath MA, et al. Pelvic organ prolapse and collagen-associated disorders. International UrogynecologyJournal [internet] 2012[citado 2012 mar]; 23(3):[Aprox.6p].Disponible en : https://www.ncbi.nlm.nih.gov/pubmed/21811768
24. GarcíaCampayo J, Asso E, Alda M, Andres EM, Sobradiel N. Association between joint hypermobility syndrome and panic disorder: a case-control study. Psychosomatics[internet] 2010[citado 2010 jan –feb] ;51(1):[Aprox.6p].Disponible en : http://www.sciencedirect.com/science/article/pii/S0033318210706599
25. Campo M C. Evaluación de la hemostasia en niños con síndrome de Ehlers-Danlos. Rev. Cubana Hematología, Inmunología y Hemoterapia[internet] 2014 junio[Citado 2017 jun 20] ;30(2):[Aprox.8p].Disponible en : http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892014000200007
26. Rombaut L, Scheper M, De Wandele I, De Vries J, Meeus M, Malfait F, et al. Chronic pain in patients with the hypermobility type of Ehlers-Danlos syndrome: evidence for generalized hyperalgesia. ClinRheumatol [internet] 2014 [citado 2015 jun ];34(6):[Aprox.8p].Disponible en : https://www.ncbi.nlm.nih.gov/pubmed/24487572
27. Rombaut L, Malfait F, Cools A, De Paepe A, Calders P. Musculoskeletal complaints, physical activity and health-related quality of life among patients with the Ehlers-Danlos syndrome hypermobility type. Disabil Rehabil [internet] 2010 [citado 2015 sep 21] ;32(16):[Aprox.6p].Disponible en : https://www.ncbi.nlm.nih.gov/pubmed/20156051
28. Quispe Pari G D. Sindrome de EhlersDanlos (SED). Rev. Act. Clin. Med [Internet] 2015 [citado 2015 Sep 21];45. Disponible en: http://www.revistasbolivianas.org.bo/scielo.php?script=sci_arttext&pid=S2304-37682014000600002&lng=es.
29. Pauker SP , Stoler J. Clinical manifestations and diagnosis of Ehlers-Danlos síndromes [internet].2017[citado 2017 jun 21].Disponible en : https://www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-ehlers-danlos-syndromes
30. De Wandele I, Rombaut L, Leybaert L, van de Borne P, de Backer T, Malfait F, et al. Dysautonomia and its underlying mechanisms in the hypermobility type of Ehlers-Danlos syndrome. Semin Arthritis Rheum[internet] 2014[citado 2014 ago 24];44(1):[Aprox.7p].Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/24507822
31. Linfante I, Lin E, Knott E, Katzen B, Dabus G. Endovascular repair of direct carotid-cavernous fistula in Ehlers-Danlos type IV. J NeurointervSurg[internet[ 2014[citado 2014 jan ];7(1):e3 .Disponible en : https://www.ncbi.nlm.nih.gov/pubmed/24401479
32. Rombaut L, Malfait F, De Wandele I, Taes Y, Thijs Y, De Paepe A, et al. Muscle mass, muscle strength, functional performance, and physical impairment in women with the hypermobility type of Ehlers-Danlos syndrome. ArthritisCare Res (Hoboken) [internet]2012[citado 2012 oct ] ;64(10):[Aprox.8p].Disponible en : https://www.ncbi.nlm.nih.gov/pubmed/22556148
33. Danese C, Castori M, Celletti C, et al. Screening for celiac disease in the joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type. American Journal of Medical Genetics A [internet] 2011[citado 2011 sep]; 155A(9):[Aprox.2p].Disponible en : https://www.researchgate.net/publication/51545908_Screening_for_Celiac_Disease_in_the_Joint_Hypermobility_SyndromeEhlers-Danlos_Syndrome_Hypermobility_Type
34. Keer R, Simmonds J. Joint protection and physical rehabilitation of the adult with hypermobility syndrome. Current Opinion in Rheumatology[internet] 2011[citado 2011 mar ];23(2):[Aprox.5p]. Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/21252682
35. Kanjwal K, Saeed B, Karabin B, Kanjwal Y, Grubb BP. Preliminary observations suggesting that treatment with modafinil improves fatigue in patients with orthostatic intolerance. American Journal of Therapeutics [internet] 2011 [citado 2011 nov ] ;18(6):[Aprox.3].Disponible en : https://www.ncbi.nlm.nih.gov/pubmed/20393343
36. Eccles JA, Beacher FD, Gray MA, et al. Brain structure and joint hypermobility: relevance to the expression of psychiatric symptoms. The British Journal of Psychiatry[internt] 2012 [citado 2012 jun] ; 200(6):[Aprox.1p].Disponible en : https://www.ncbi.nlm.nih.gov/pubmed/22539777
37. Refai H, Altahhan O, Elsharkawy R. The efficacy of dextrose prolotherapy for temporomandibular joint hypermobility: a preliminary prospective, randomized, double-blind, placebo-controlled clinical trial. Journal of Oral and MaxillofacialSurgery [internet] 2011[citado 2011 dic];69(12):[Aprox.8p].Disponible en : https://www.ncbi.nlm.nih.gov/pubmed/21757278
Marianela Puerto Martínez: Médica. Especialista de Primer Grado en Pediatría. Policlínico Universitario Manuel Pity Fajardo. Guane. Cuba. Si usted desea contactar con el autor de la investigación hágalo aquí

