










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La enfermedad de Creutzfeldt-Jakob (CJD) es un trastorno neurodegenerativo causado por priones en humanos. Se presenta como encefalopatía espongiforme de distribución mundial con una prevalencia de 1 cada 1 000 000 de habitantes. Los priones pueden colonizar diferentes tejidos, pero el único sistema en el cual se ha demostrado daño histopatológico, tanto en animales como en humanos, es el sistema nervioso central.1
La enfermedad tiene tres tipos etiológicos: esporádica, hereditaria y adquirida. La patogénesis molecular de todos los tipos implica el plegamiento incorrecto y la agregación de la proteína priónica celular (PrPC) en una serie de formas asociadas a la enfermedad. La forma más común de presentación es esporádica, representa alrededor del 85 % de los casos reportados. Por lo general, ocurre en edades avanzadas, con un promedio de 67 años, acompañada de una corta supervivencia pos-diagnóstico de alrededor de cuatro meses.2 Clínicamente el CJD se caracteriza por el desarrollo de una demencia rápidamente progresiva asociada con ataxia mioclónica,3 a medida que avanza la enfermedad el daño neuronal progresivo debido a los depósitos de PrP conduce a un deterioro mental más pronunciado, a la aparición de trastornos del movimiento, ceguera y coma.4
Se requiere la confirmación de la autopsia o biopsia cerebral para obtener un diagnóstico definitivo de la enfermedad. La identificación del subtipo o variante de CJD depende de la identificación correcta de la fuente de contaminación a la que los pacientes han estado expuestos, así como los factores de riesgo que presenten.2 Se han desarrollado una variedad de herramientas de diagnóstico recientes que permiten un diagnóstico ante mortem más confiable de la enfermedad por priones, como la resonancia magnética del cerebro y cerebrospinal fluid real-time quaking-induced convertion, que han permitido un mejor manejo de los casos diagnosticados a pesar de la letalidad de la enfermedad, siendo pruebas importantes que se deben utilizar al evaluar a pacientes con sospecha de CJD.5
En Cuba, la incidencia de la enfermedad es casi nula, sin embargo, tras resultados arrojados por exámenes de anatomía patológica a pacientes con sospecha de la misma se han diagnosticado en todo el país un número significativo de casos de etiología no hereditaria. Por dicha cuestión, el objetivo principal de esta investigación es caracterizar la enfermedad de Creutzfeldt-Jakob en el Instituto Nacional de Neurología y Neurocirugía durante el periodo de febrero 1981 a enero 2019.
MÉTODOS
Se realizó un estudio observacional, descriptivo transversal en pacientes diagnosticados con enfermedad de Creutzfeldt-Jakob, de etiología no hereditaria, en el Instituto nacional de Neurología y Neurocirugía de Cuba entre 1981 y 2019.
El universo estuvo constituido por los 12 pacientes diagnosticados en dicho periodo. El criterio de inclusión fue el diagnóstico de la enfermedad mediante la autopsia y la ausencia de antecedentes patológicos familiares relacionados con la enfermedad. La información recogida se obtuvo a partir de las Historias Clínicas de los pacientes seleccionados.
Las variables estudiadas fueron: edad; sexo; raza (blanca, mestiza, negra); estadía hospitalaria (días), ocupación, antecedentes patológicos personales, primer síntoma identificado, hallazgos radiológicos, resultados de química sanguínea (normal, patológico), electroencefalograma (normal, patológico), resultados del estudio del líquido cerebroespinal (normal, patológico), manifestaciones clínicas.
Los datos obtenidos se procesaron en una base de datos en Microsoft Excel®. Los datoscuantitativosse procesaroncon el sistema estadístico SPSS. Se utilizaron técnicas de estadística descriptiva, los resultados obtenidos se expresaron como frecuencias absolutas y relativas porcentuales.
Para la realización de la investigación se tuvieron en cuenta los principios éticos establecidos en la Declaración de Helsinki. Como parte de la investigación se garantizó la confidencialidad de todos los datos obtenidos, los cuales fueron empleados con fines estrictamente investigativos.
RESULTADOS
En el estudio realizado fueron analizados los datos de 12 pacientes en los que se encontró una edad promedio de 60 años con una edad mínima de 48 y máxima de 79 años, para una desviación estándar de 8,6 años. Se identificó un predominio del sexo femenino que representó el 66,3 %.
Los pacientes de raza blanca representaron el 83,3 % mientras los de raza negra el 16,4 %. La estadía hospitalaria media fue de 9,8 días con una desviación estándar de 11 días para un mínimo de siete y un máximo de 46 días.
La ocupación más frecuente fue la de jubilado para un 41,6 % seguida de la ocupación ama de casa que representó el 16 %. El antecedente patológico personal más frecuente fue la hipertensión arterial, que representó el 66,6 %.
El primer síntoma identificado por el paciente que se presentó con mayor frecuencia fue la dificultad para caminar (50 %) seguido de la pérdida del equilibrio (33 %) y los mareos (17 %). (Fig. 1)
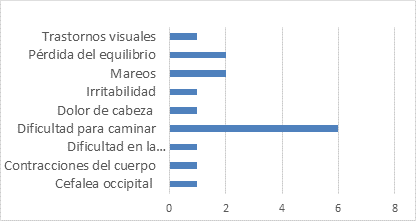
El hallazgo radiológico predominante fue la atrofia frontoparieto temporal y cerebelosa en el 50 % de los pacientes. En el 83,3 % de los pacientes se obtuvieron resultados de química sanguínea normales. El 41,6 % presentó electroencefalograma patológico. En el 50 % se identificaron valores anormales en el líquido cerebroespinal.
Las manifestaciones clínicas más frecuentes fueron la afasia en un 25 %, desorientación, disartria, disfagia y los trastornos de la micción en la misma proporción. (Tabla 1)
Tabla 1 Manifestaciones clínicas en pacientes diagnosticados con enfermedad de Creutzfeldt-Jakob
| Manifestaciones clínicas | Fa | Fi |
|---|---|---|
| Afasia | 3 | 25 |
| Desorientación | 3 | 25 |
| Disartria | 3 | 25 |
| Disfagia | 3 | 25 |
| Trastornos de la micción | 3 | 25 |
| Amnesia | 2 | 16,7 |
| Insomnio | 2 | 16,7 |
| Parestesia | 2 | 16,7 |
| Temblores generalizados | 2 | 16,7 |
| Trastornos de la marcha | 2 | 16,7 |
| Alucinaciones | 1 | 8,33 |
| Alucinaciones | 1 | 8,33 |
| Cefalea | 1 | 8,33 |
| Confusión | 1 | 8,33 |
| Descuido del autocuidado | 1 | 8,33 |
| Disfunción eréctil | 1 | 8,33 |
| Pérdida del equilibrio | 1 | 8,33 |
Fuente: Base de datos confeccionada
DISCUSIÓN
Los resultados de este estudio mostraron un predominio de edades por encima de los 48 años con una media de 60. Estos hallazgos coinciden con un registro realizado en China, 6 y en una investigación realizada en por Chang Qi et al.7) Esta última recogió además, datos de pacientes en edades entre 20 y 50 años sin antecedentes genéticos de la enfermedad demostrables, en los pacientes más jóvenes el tiempo de supervivencia fue mayor.
La mayoría de estudios anteriores muestran una mayor incidencia de la enfermedad en el sexo femenino, lo cual se corresponde con los resultados encontrados.6,7,8
En este estudio hubo un alto predominio de los pacientes de raza blanca en relación a la raza negra. Un estudio estadístico realizado en los Estados Unidos,8 mostró resultados muy similares asociados a los porcientos demográficos de la población norteamericana similares a los de Cuba, 73 % blancos - 12,7 % negros y 65,05 % blancos - 10,08 % negros, respectivamente.8,9
Por la relación de estos percentiles en la población general y en los pacientes estudiados estos datos se mostraron poco relevantes. No existen datos específicos que relacionen la ocupación de los pacientes con la enfermedad, este estudio mostró mayor incidencia en pacientes no vinculados a una actividad laboral con un predominio de personas jubiladas y amas de casa.
Según los resultados descritos por Hermann et al. (10 se encontró la posibilidad de un aumento de la tasa de médicos entre todos los casos, otro estudio reportó un caso de un técnico con una variante de CJD, relacionado posiblemente con su ocupación al haber manipulado, años atrás, muestras murinas contaminadas con el agente que causa encefalopatía espongiforme bovina.11
Hasta el momento no se han establecido antecedentes patológicos personales específicos para la aparición de Creutzfeldt-Jakob esporádico. Investigaciones anteriores sugieren la posibilidad de transmisión en procederes iatrogénicos fundamentalmente quirúrgicos. Según un análisis realizado por Martheswaran et al,12) se encontraron 10 casos con transmisión iatrogénica luego de un trasplante de córnea, asociado a órganos infectados donde predominaron como factores de riesgo la edad avanzada de los donantes y un historial de trasplantes múltiples en los receptores.
En este sentido, el actual estudio mostró un predominio de la hipertensión arterial (HTA) dentro de los antecedentes más frecuentes encontrados en los pacientes estudiados. Esto coincide con un caso reportado por Qavi et al.,13 de un paciente masculino de 67 años con el antecedente de hipertensión arterial sin referencias de viajes, contacto con animales salvajes o síntomas similares entre contactos cercanos. En Cuba la HTA es uno de las patologías más frecuentes en la población adulta mayor; La Habana es una de las provincias con mayor tasa de prevalencia. 14 Estos datos pudieran relacionarse con los hallazgos del estudio, lo que aumenta la posibilidad de que estos resultados se asocien a proporciones estadísticas y no específicamente por una relación directa con la CJD.
La mayor parte de la literatura coincide en que, dentro de los primeros síntomas que se identifican en los pacientes es una alta incidencia de demencia y pérdida de memoria seguido por manifestaciones cerebelosas.6,7,13,14 Los resultados obtenidos mostraron un mayor por ciento de manifestaciones cerebelosas que incluyeron dificultad para caminar, pérdida del equilibrio y mareos. Los síntomas asociados a la pérdida de memoria no aparecieron reflejados en las historias clínicas y los datos de los pacientes estudiados, por lo que esta variable no pudo ser estudiada. Es probable que con la progresión de la enfermedad a fases más avanzadas, los síntomas psiquiátricos están disminuidos o son menos notables debido al resto de manifestaciones que serían mejor percibidas.15
Estos síntomas se relacionan con los hallazgos radiológicos encontrados en la mitad de los pacientes del estudio, correspondientes a una atrofia fronto - parieto - temporal y cerebelosa. Existen hipótesis que indican que la transformación de las proteínas priónicas en priones, así como la acumulación de priones, conducen a la neurodegeneración.16 Según hallazgos de Younes et al.,17 la atrofia cerebral localizada aparece como una característica en los pacientes con CJD, tanto en evoluciones progresivas como los de rápida instauración, donde se observó atrofia regional selectiva en una combinación de regiones de núcleos corticales y profundos, predominante en las regiones de asociación multimodal además de cortezas prefrontales laterales e inferiores bilaterales y tálamos bilaterales.
Un elevado por ciento de los pacientes estudiados presentó anormalidades en el líquido cerebroespinal. La prueba RT-QUIC (Real-Time QUaking-Induced Conversión) de segunda generación, es muy sensible y específica para la CJD, detecta proteínas priónicas patógenas en el líquido cefalorraquídeo de los pacientes.16 Los registros estudiados no contaban con este estudio por lo que no se evidenció la relación entre esta variable y el diagnóstico o evolución de la enfermedad. Un estudio realizado por Daniel D Rhoads et al., 18 mostró las limitaciones de sensibilidad asociadas al diagnóstico de CJD a partir de esta prueba, demostró la eficacia del RT-QuIC para mejorar, en gran medida, la detección de enfermedades priónicas en el laboratorio con fines diagnósticos.
Las manifestaciones clínicas halladas en los pacientes no siguieron patrones específicos, se encontró un predominio de la afasia, desorientación, disartria y los trastornos de la micción en el mayor por ciento de los pacientes. Resultados similares se encontraron en un caso presentado por Mahboob et al.,19 y otras investigaciones realizadas.8,13
Es importante mencionar que el presente trabajo tiene como principal limitación que el número de pacientes con CJD en el estudio es reducido, por lo que la representación y alcance de los datos es baja.
En la investigación realizada se observó un mayor número de casos del sexo femenino. La dificultad para caminar seguido de la pérdida del equilibrio y los mareos, fueron los primeros síntomas identificados. Predominó como hallazgo radiológico, la atrofia frontoparietotemporal y cerebelosa. Las manifestaciones clínicas más frecuentes fueron: la afasia, desorientación, disartria, disfagia y los trastornos de la micción.

