










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Médica Electrónica
versión On-line ISSN 1684-1824
Rev. Med. Electrón. v.31 n.4 Matanzas jul.-ago. 2009
HOSPITAL PEDIÁTRICO DOCENTE PROVINCIAL ELISEO NOEL CAAMAÑO. MATANZAS
Anomalía de Pelger Hüet. A propósito. de un caso
Pelger-Hüet anomaly. Apropos of a case
AUTORES
Dra. Adys Gutiérrez Díaz. (1)
E-mail:adysg.mtz@infomed.sld.cu
Lic. Lourdes Díaz Naranjo. (2)
Dr. Luis Ramón Rodríguez. (3)
Dr. Juan Ramírez Díaz. (4)
Téc. Elizabeth Suárez García.(5)
Dr. Guillermo Montalván González.(6)
1. Especialista I Grado Hematología. Profesora Instructora. Hospital Pediátrico Docente "Eliseo Noel Caamaño".Matanzas
2. Licenciada en Tecnología de la Salud. Profesora Instructora. Hospital Pediátrico Docente "Eliseo Noel Caamaño".Matanzas
3. Especialista de I Grado Hematología. Especialista de I Grado Medicina General Integral. Instituto Nacional de Hematología e Inmunología.La Habana. Matanzas
4. Especialista de I Grado Hematología. Hospital Pediátrico Docente "Eliseo Noel Caamaño".Matanzas.
5.Técnica de Laboratorio Clínico. Hospital Pediátrico Docente "Eliseo Noel Caamaño". Matanzas.
6.Especialista I Grado Pediatría. Profesor Instructor. Hospital Pediátrico Docente "Eliseo Noel Caamaño". Matanzas.RESUMEN
La anomalía de Pelger Hüet fue descrita inicialmente en 1928 por el médico holandés Pelger y su origen genético fue descubierto más tarde por el pediatra Hüet. Se transmite con un carácter autosómico dominante y consiste en una mutación del gen que codifica el receptor de la lámina B. A partir de esta mutación se producen alteraciones en el núcleo de los leucocitos, fundamentalmente en los neutrófilos, con afectación de la segmentación nuclear y trastornos en la cromatina. Presentamos a un paciente que fue ingresado en el hospital por una mordedura de animal y se describió en la lámina periférica la presencia de neutrófilos hipolobulados. El carácter familiar se confirmó por el hallazgo de esta alteración en la madre del paciente.
DeCS
ANOMALÍA DE PELGER-HUET /diagnóstico
ANOMALÍA DE PELGER-HUET /sangre
ANOMALÍA DE PELGER-HUET /genética
HUMANO
NIÑOINTRODUCCIÓN
En 1928 el médico holandés Pelger describió dos pacientes con una alteración morfológica de los leucocitos que consistía en una hipolobulación del núcleo, con una cromatina más densa y gruesa. Esta anomalía fue descrita más adelante como una alteración genética por el pediatra Hüet. En años posteriores se reportaron varias familias con la Anomalía de Pelget Hüer (APH) como una alteración benigna autosómica dominante, con afectación de la forma nuclear y la distribución de la cromatina en los granulocitos, sin afectar su función. En las diferentes investigaciones realizadas la incidencia de este desorden varía desde un rango tan alto como 1 en 1000 personas, hasta rangos de 1 en 4000, 6000 o aún en 10 000 personas. Originalmente observada sobre todo en Holanda, Alemania y Suiza, la anomalía fue descrita posteriormente en otras partes del mundo, incluyendo individuos asiáticos y descendientes de africanos. (1, 2)
La importancia práctica de determinar la APH consiste en distinguir este defecto de la desviación a la izquierda que ocurre en asociación con infecciones y otras patologías, además de diferenciarla de la anomalía de pseudopelger Hüet presente en otras situaciones clínicas (3). Se describe un caso de un paciente pediátrico que ingresa por mordedura de animal (equino) en la mejilla izquierda, y que en los complementarios realizados se detectó esta alteración morfológica.Caso Clínico
Paciente de 7 años de edad, blanco, masculino, con antecedentes de salud y procedente de un área rural. Ingresa por una mordedura de animal (equino) en la mejilla izquierda. Al examen físico solo presentaba laceración de toda la mejilla izquierda.
Complementarios realizados:
ALAT: 9 UI, ASAT: 7U/I, Creatinina: 53 µmol/l; Proteínas totales: 73g/l, Albúmina: 48g/l, glicemia: 4,3mmol/l.
Estudios hematológicos: Hb: 12.1 g/dl, Hto: 0.39, plaquetas: 300×10 9 /l, leucocitos: 12.3×10 9 /l, neutrófilos: 78%, linfocitos: 17%, monocitos: 3%, eosinófilos: 2%, reticulocitos: 0.9%, eritrosedimentación: 15 mm/h.
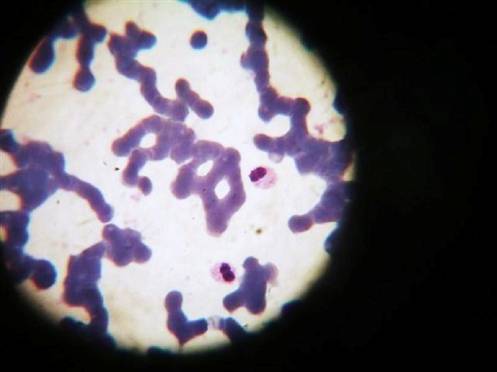
Lámina periférica: Normocitosis, normocromía, ligera leucocitosis con neutrofilia, neutrófilos hipolobulados: 27% monolubulados y 63% bilobulados, plaquetas adecuadas.Figura No.1
Lámina periférica de la madre: Normocitosis, normocromía, leucocitos normales con neutrófilos hipolobulados: 31% monolubulados y 62% bilobulados, plaquetas adecuadas.
DISCUSIÓN
En la APH la gran mayoría de los casos descritos son heterocigóticos y es más frecuente encontrar neutrófilos con formas bilobuladas del núcleo, en forma de gafas o anteojos, y la forma de vara o vástago. El 69 a 93% de los neutrófilos deben ser hipolobulados, pocas células contienen tres lobulaciones (usualmente menos del 10%) y es raro encontrar células con 4 lobulaciones. En los casos homocigóticos hay un predominio de núcleo redondo sin evidencia de segmentación (90 a 100% de los neutrófilos). En esta anomalía la cromatina es más densa y gruesa y la regularidad del núcleo contrasta con la irregularidad de los lóbulos vistos en los neutrófilos normales. En un examen de lámina periférica normal menos del 27% de las células son bilobuladas y un número significativo de células tienen 3 o más lobulaciones. Un incremento en el número de neutrófilos lobulados se describió en una paciente con esta anomalía, que desarrolló una anemia perniciosa. La sobrevida de las células con APH en la circulación es normal al igual que su función, y son capaces de destruir y fagocitar microorganismos (1-3). La presencia de alteraciones similares en la sangre periférica de otros miembros de la familia ayuda a establecer el diagnóstico.
El carácter homocigótico para la APH se describió primeramente en conejos. Desde entonces se ha asociado con anomalías esqueléticas y alta mortalidad, basado fundamentalmente en estudios hechos en animales (1,2). En 1952 Haverkamp Begeman y colaboradores fueron los primeros en describir la APH homocigótica en una niña holandesa que padecía de convulsiones y tenía un discreto retardo en el desarrollo psicomotor. A pesar de presentar una baja talla, no se advirtieron anomalías esqueléticas. Posteriormente se describieron otros casos en Marruecos, Rumania, Italia, España y Alemania. Algunos de los pacientes presentaron defectos esqueléticos dentro de los cuales se describen polidactilia y acortamiento de los huesos metacarpianos. Por lo general los casos pueden alcanzar la vida adulta. (2-5)
Las alteraciones genéticas responsables de la APH se describieron recientemente. Estudios genómicos muestran que la APH está ligada al cromosoma 1 q41_43. En este cromosoma se lograron identificar alteraciones en la secuencia del gen del receptor de la lámina B (LBR), dando lugar a la ausencia de la proteína LBR, miembro de la familia esterol reductasa, integrada estrechamente a la membrana nuclear cuya función es unir a la heterocromatina y a las láminas con la membrana nuclear. Las células heterocigóticas afectadas con la APH muestran una expresión reducida del receptor de la lámina B y las células homocigóticas contienen solo trazas del mismo. Se ha demostrado que la expresión del receptor de la lámina B afecta la forma nuclear y la distribución de la cromatina nuclear. (3,4,6-10)
La APH se ha descrito además en otras células como los eritroblastos, monocitos, linfocitos, células plasmáticas, eosinófilos y basófilos, las cuales presentan una hipercondensación de la cromatina. Un tercio de los megacariocitos son también binucleados como los neutrófilos. (1,6)
La llamada anomalía de pseudopelger Hüet ha sido descrita en las Leucemias Agudas no Linfoblásticas, la Leucemia Mielode Crónica y los Síndromes Mielodisplásticos. Puede estar presente además en el mixedema asociado a panhipopituitarismo, el mieloma múltiple, reacción leucemoide en las metástasis de médula ósea, infecciones enterovirales, distrofia muscular, en el déficit de ácido fólico y vitamina B12 y en la sensibilidad a drogas (3). Comparada con la APH, en la anomalía de pseudopelger el patrón de la lobulación de los neutrófilos y la densidad de la cromatina es más heterogéneo. El porcentaje de neutrófilos hipolobulados es menor, lo que contrasta con un mayor número de neutrófilos trilobulados. La ausencia de transformación a una leucemia aguda o un síndrome mielodisplástico en la anomalía familiar, contrasta con la naturaleza neoplásica de la anomalía de pseudopelger Hüet. (3,6)
El caso que presentamos constituye un ejemplo más de APH, donde la experiencia del personal y el estudio familiar influyó en su detección. Aunque la entidad homocigótica es rara, el consejo genético pudiera prevenir su aparición en la descendencia de este paciente. Nosotros, los técnicos y hematólogos debemos estar alertas y bien preparados para detectar estas alteraciones congénitas y reportar nuestras observaciones de cualquier anomalía morfológica desconocida.REFERENCIAS BIBLIOGRÁFICAS
1. Keith M Skubitz. Qualitative Disorders of Leukocytes. Wintrobe's Clinical Hematology. 11th ed. USA: Lippincott Willians and Wilkins Publishers ;2003
2. Oosterwijk JC, Mansour S, Van Noort G, Waterham H, Hall C, Hennekam R. Congenital abnormalities reported in Pelger-Huët homozygosity as compared to Greenberg/HEM dysplasia: highly variable expression of allelic phenotypes. J Med Genet.2003; 40:937-41.
3. Kanwar V, Shafer F. Pelger-Huet Anomaly; 2007. Consultado 17 de sept.2007. Http:// emedicine.medscape.com.
4. Hoffmann K, Dreger CK, Olins AL, Olns DE, Shultz LD, Lucke B. Mutations in the gene encoding the lamin B receptor produce an altered nuclear morphology in granulocyte. Nat Genet. 2002 Aug; 31(4):410-4[ STANDARDIZEDENDPARAG]
5. Waterham H, Koster J, Mooyer P, Van Noort G, Kelley R, Wilcox W, et al. Autosomal Recessive HEM/Greenberg Skeletal Dysplasia Is Caused by 3 ß -Hydroxysterol ? Reductase Deficiency Due to Mutations in the Lamin B Receptor Gene. Am J Hum Genet. 2003 April; 72(4): 1013–7[ STANDARDIZEDENDPARAG]
6. Tomonaga M. Nuclear abnormalities in Pelger-Huet anomaly; progress in blood cell morphology. Rincho Byori.2005 Jan; 53(1):54-60.
7. Best S. Lamin B-receptor mutations in Pelger-Huet anomaly. Br J Haematol. 2003 Nov;123(3):542-4.
8. Gruenbaum Y. The nuclear lamina and its functions in the nucleus. Int Rev Cytol. 2003; 226:1-62.
9. Sjakste N, SjaksteT. Nuclear matrix proteins and hereditary diseases. Genetika. 2005 Mar; 41(3):293-8
10. Kalfa TA, Zimmerman SA , Goodman BK , McDonald MT, Ware RE. Pelger-Huet anomaly in a child with 1q42.3-44 deletion. Pediatr Blood Cancer. 2006 May 1; 46(5):645-8.
SUMMARY
The Pelger-Hüet anomaly was described firstly in 1928 by the Holland physician Pelger and its genetic origin was discovered later by the pediatrician Hüet. It is transmitted with a dominant autosomal character and it is a mutation of the gene codifying the plate B receptor. Beginning from this mutation, the alteration of white cells core took place, mainly in neutrophils, with affectations of the nuclear segmentation and chromatin disturbances. We present a patient entering our hospital as a cause of an animal bite, and there were discovered hypolobulated neutrophils in the periferal plate. The familiar character was confirmed with the finding of this alteration in the patient's mother.
MeSH
PELGER-HUET ANOMALY / diagnosis
PELGER-HUET ANOMALY /blood
PELGER-HUET ANOMALY /genetics
HUMANS
CHILDCÓMO CITAR ESTE ARTÍCULO
Gutiérrez Díaz A, Díaz Naranjo L, Ramón Rodríguez L, Ramírez Díaz J, Suárez García E, Montalván González G. Anomalía de Pelger Hüet. A propósito de un caso. Rev méd electrón[Seriada en línea] 2009; 31(4). Disponible en URL: http://www.revmatanzas.sld.cu/revista%20medica/año%202009/vol4%202009/tema16.htm [consulta: fecha de acceso]

