










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión impresa ISSN 0034-7515
Rev Cubana Farm vol.47 no.1 Ciudad de la Habana ene.-mar. 2013
ARTÍCULOS ORIGINALES
Control de la calidad y estudio de estabilidad del paracetamol gotas orales 100 mg/ml
Quality control and stability study of 100 mg/ml paracetamol oral drops
MSc. Caridad M. García Peña, Tec. Yanet Montes de Oca Porto, MSc. Suslebys Salomón Izquierdo
Centro de Investigación y Desarrollo de Medicamentos. La Habana, Cuba
RESUMEN
Introducción: las gotas orales de Paracetamol, están indicadas a la población infantil hasta los 5 años para el alivio de la fiebre, dolor de cabeza, dolores dentales y proporciona alivio sintomático del resfriado común.
Objetivo: validar dos métodos analíticos, para el control de la calidad y el estudio de estabilidad y estudiar la estabilidad de las gotas orales de producción nacional.
Métodos: para cuantificar el principio activo para el estudio de estabilidad, la separación se realizó a través de una columna cromatográfica Lichrosorb RP - 18 (5µm) (250 x 4 mm), con detección ultravioleta a 243 nm, empleando una fase móvil compuesta por Agua destilada: Metanol (3:1). Mientras que el método para el control de la calidad se utilizó un Espectrofotómetro SPECTRONIC GENESYS 2. Para el estudio de estabilidad, se emplearon los métodos de vida de estante (a temperatura inferior a 30 o C) y de estabilidad acelerada (40 ± 2ºC) mediante cromatografía líquida de alta eficiencia.
Resultados: los resultados obtenidos de los parámetros evaluados en las validaciones se encontraron dentro de los límites establecidos. Los resultados del estudio de estabilidad realizado, demuestran que el producto terminado cumplió con las especificaciones de calidad durante el estudio.
Conclusiones: los métodos analíticos por espectrofotometría UV y cromatografía líquida de alta resolución, son válidos para el control de la calidad y estudio de estabilidad de las gotas orales de Paracetamol 100 mg/mL, ya que resultaron lineales, precisos, exactos y específicos. Se demostró la estabilidad física, química y microbiológica del producto por espacio de 12 meses a temperatura inferior a 30 ºC, envasados en frascos de vidrio ámbar por 15 mL, boca 18 mm, calidad hidrolítica III. Además se evidenció que el producto es estable durante 30 días después de abierto el frasco.
Palabras clave: Paracetamol, Espectrofotometría, Cromatografía Líquida de Alta Resolución, validación, estabilidad acelerada, estabilidad por vida de estante.
ABSTRACT
Introduction: Paracetamol is an effective analgesic and antipyretic drug of the non-steroidal anti-inflammatory drug group. Paracetamol oral drops are indicated for use in infant population aged up to 5 years to relieve fever, headache, toothache and symptomatic relief of common cold.
Objective: to validate two analytical methods for the quality control and the stability study and to study the stability of 100 mg/ml Paracetamol oral drops made in Cuba.
Methods: for quantification of the active principle in the final product in order to study stability, a chromatographic column equipment called Lichrosorb RP-18 was used for separation (5µm) (250 x 4 mm), with ultraviolet ray detection at 243 nm and a mobile phase made up of distilled water: methanol (3:1) and the quantification of this principle against a reference sample by using the external standard method. For the quality control, the spectrophotometry used the spectrophotometer SPECTRONIC GENESYS 2, the ideal wavelength was 245 nm since it matches the maximum absorption rate and there is no interference with the excipients. As to the stability study of the drops, the on- shelf life method (temperature below 30 C) was used, and for the accelerated stability analysis (40 ± 2ºC) through high performance liquid chromatography.
Results: the results of the evaluated parameters in the validation of the methods for the quality control and the stability study were within the set limits. The results of the stability study, both accelerated and on- shelf life and reservoir use, showed that the final product met the quality specifications during the study.
Conclusions: the analytical methods based on UV spectrophotometry and high performance liquid chromatography are valid for the quality control and the stability study of 100 mg/ml Paracetamol oral drops, since they are linear, precise, accurate and specific. The physical, chemical and microbiological stability of the product was proved for 12 months at a temperature below 30ºC, packed in 15 ml amber glass reservoirs, 18mm opening and hydrolytic quality III. The product is also stable for 30 days after opening the reservoir.
Keywords: Paracetamol, spectrophotometry, high performance liquid chromatography, validation, accelerated stability, on-shelf life.
INTRODUCCIÓN
Las gotas orales de Paracetamol, están indicadas a la población infantil hasta los 5 años para el alivio de la fiebre, dolor de cabeza, dolores dentales y proporciona alivio sintomático del resfriado común.
El Paracetamol es un analgésico y antipirético eficaz, perteneciente al grupo de los AINES (antiinflamatorios no esteriodeos). Sus efectos terapéuticos, similares a las de los salicilatos, están relacionados con la inhibición de la síntesis de prostaglandinas, como resultado de la disfunción de la ciclooxigenasa. Su acción analgésica es debido a la elevación del umbral del dolor y antipirética por su acción sobre el centro regulador del calor en el hipotálamo. Es un antiinflamatorio débil, debido a que los tejidos inflamados poseen niveles más altos de peróxidos que impiden que la ciclooxigenasa sea inhibida por el Paracetamol. Sus acciones son iguales en efectividad que las del Ácido acetilsalicílico pero sin los efectos adversos asociados a este último.
Las gotas orales de Paracetamol 100 mg/mL, deben administrarse cada 4 a 6 horas sin exceder 5 dosis en un período de 24 horas, no más de 60 mg/ Kg de peso en 24 horas. 1-2
Los métodos espectrofotométricos resultan de gran utilidad para el control de la calidad porque son rápidos, sencillos y económicos, mientras que los métodos cromatográficos son más selectivos y específicos para los estudios de estabilidad del producto terminado y permiten el monitoreo del principio activo en el tiempo. 3-4
La estabilidad de los productos farmacéuticos representa un importante eslabón en el desarrollo y formulación de toda forma terminada. De esta manera se puede definir las condiciones de almacenamiento en el envase propuesto y establecer el tiempo de vida útil. Estos estudios contemplan la conservación de la potencia, pureza, características organolépticas y su efectividad.5-6
La estabilidad de un medicamento puede verse afectada por diversos factores como son las condiciones ambientales como: luz, humedad, temperatura y aire, factores intrínsecos a la fabricación: tamaño de partícula, pH, naturaleza del envase y presencia de otros productos químicos procedentes de contaminación.7-8
Los objetivos de este trabajo fueron validar dos métodos analíticos, para el control de la calidad y el estudio de estabilidad y estudiar la estabilidad de las gotas orales de Paracetamol 100 mg/mL de producción nacional.
MÉTODOS
La sustancia de referencia química de Paracetamol fue suministrada por el grupo de sustancias de referencia del Centro de Investigación y Desarrollo de Medicamentos (CIDEM, La Habana, Cuba), el cual fue analizado por el método cromatográfico establecido para realizar el control de la calidad de la materia prima, con una pureza de 99,8 %.9
El presente estudio de las gotas orales de Paracetamol 100 mg/mL de producción nacional, se realizó por los métodos de vida de estante y estabilidad acelerada. Se emplearon muestras de los lotes 9001, 9002 y 9003, elaborados con el lote de Paracetamol (materia prima) 0711045, procedente de la China, producidos en la Empresa Laboratorio Farmacéutico Líquidos Orales de Bayamo, MEDILIP y envasados en frascos de vidrio ámbar por 15 mL, boca 18 mm, calidad hidrolítica III, con tapa plástica, blanca, de 18 mm de diámetro interior, con reductor gotero de 18 mm de diámetro exterior adecuado al frasco y al producto, roscada, con anillo de inviolabilidad.
Validación de los métodos de análisis
Todos los reactivos utilizados fueron de calidad analítica, procedentes de la Riedel de Haen.
El producto terminado en forma de gotas orales empleado en el estudio de validación de los métodos fue el lote 9001, el cual cumplió con las especificaciones de calidad establecidas para el control de la calidad de las gotas orales.
Se realizó la validación de dos métodos de análisis de las gotas orales de producción nacional de Paracetamol, uno por Cromatografía Líquida de Alta Resolución para estudio de estabilidad y el otro por Espectrofotometría para el control de la calidad.
Para el estudio de la validación del método del estudio de estabilidad, se utilizó un cromatógrafo líquido con una bomba Knauer, detector UV/VIS (Knauer) ajustado a 243 nm, un inyector con un rulo de 20 µl e integrador (SHIMADZU CR 8 A). La separación se realizó isocráticamente con una columna Lichrosorb RP - 18 (5µm) (250 x 4 mm). La fase móvil empleada, consistió en una mezcla de Agua destilada: Metanol (3:1), a un flujo de 1,5 mL/min.9
En el caso del método para el control de la calidad por espectrofotometría se utilizó un Espectrofotómetro SPECTRONIC GENESYS 2, seleccionándose 245 nm como la longitud de onda idónea para el trabajo por corresponderse con el máximo de absorción y no existir interferencias de los excipientes. Las muestras se diluyeron en la mezcla agua destilada: metanol (3:1) de modo que se obtuviera una concentración de Paracetamol de 10 µg/mL, de manera similar se preparó la sustancia de referencia química. Como blanco se empleó la mezcla agua destilada: metanol (3:1).
Los estudios de especificidad se diseñaron de forma diferente para cada método, en el espectrofotométrico se estudiaron las posibles interferencias de las sustancias auxiliares, a una concentración de 10mg/mL de muestras de producto terminado y de sustancia de referencia química y en el cromatográfico se sometieron muestras a condiciones drásticas de degradación: Hidrólisis ácida HCL 3 N, Hidrólisis básica NaOH 3 N, muestra a temperatura de 70 o C, para evaluar las posibles interferencias de los productos de degradación. 6,10
La linealidad y la exactitud, se estudiaron de forma simultánea para ambos métodos, para esto se prepararon muestras con diferentes concentraciones de Paracetamol, en el estudio de linealidad se trabajó en el intervalo de concentraciones comprendido entre el 50 al 150 % (50, 80, 100, 120 y 150 %) de la concentración teórica del principio activo, mientras que para el estudio de exactitud se emplearon tres valores de concentración (80, 100 y 120 %), realizándose los análisis por triplicado. El procesamiento estadístico de los datos se realizó por el programa Minitab 14.0, determinándose la ecuación de la recta, el coeficiente de correlación, los coeficientes de variación de los factores de respuesta y ensayo de proporcionalidad. Mientras que para el estudio de exactitud, se realizó la prueba de Cochran con vistas a comprobar si la variación de la concentración producía diferencias significativas en los resultados y la prueba de t-Student para determinar si existían diferencias significativas entre la recuperación media y el 100%.8-9
En el caso del ensayo de precisión se realizaron los estudios de repetibilidad y de precisión intermedia, trabajando a partir de una muestra homogénea común. Para el caso de la repetibilidad se trabajó sobre la base de seis determinaciones y el estudio de precisión intermedia haciendo las valoraciones por dos analistas, en tres días y a tres concentraciones (80, 100, 120 %) diferentes. A los datos se le aplicaron la prueba de t de Student y la prueba de Fischer con el objetivo de determinar si existían diferencias significativas entre las medias obtenidas y entre las precisiones alcanzadas por los analistas respectivamente, para un límite de confianza de un 95% de la media. 8-9
Estudio de estabilidad
En los estudios de estabilidad acelerada, se almacenaron las muestras de los lotes estudiados en un horno a temperatura controlada de 40 ± 2ºC y 70 ± 5 % de humedad relativa, se valoraron al inicio, 1, 2, 3 y 6 meses.
En el estudio de estabilidad por vida de estante, los lotes estudiados se almacenaron a temperatura inferior de 30 ºC, controlada diariamente registrándose valores de 26 a 29 ºC; se valoraron al inicio, a 3, 6, 9 y 12 meses de fabricados. En el estudio de estabilidad de frasco en uso, el lote estudiado (No. 9001) se almacenó a temperatura inferior de 30 ºC y protegido de la luz; el frasco se destapaba todos los días por un espacio de 3 min, simulándose la administración del medicamento por parte de los pacientes y se valoraron al inicio, a 6, 12, 18, 24 y 30 días de iniciado el estudio.
La determinación del conteo microbiológico de las gotas orales en el estudio en frasco en uso se realizó, al inicio y al final del estudio (30 días); mientras que en el estudio de estabilidad por vida de estante se realizó al inicio y al final del estudio (12 meses), empleando el método general para la realización del conteo microbiológico, reportado en la USP 32, 2009.
Para el procesamiento estadístico de los datos se hizo un análisis descriptivo en el cual se calcularon la media y la desviación estándar para cada muestra. Se aplicó además el ensayo de normalidad (Anderson-Darling) y el ensayo de homogeneidad de varianza de Levene. Ensayo de comparación múltiple de medias de Student-Newman-Keuls, para determinar diferencias entre grupos. El nivel de significación estadística, empleado en todos los casos, fue como mínimo de p < 0,05. Los datos fueron procesados utilizando el paquete estadístico MINITAB, versión 14.0.
RESULTADOS
Validación de los métodos de análisis
A la longitud de máxima absorción (245 nm) no se observa ninguna señal cuantificable atribuible a los excipientes que constituyen el placebo, a diferencia de la muestra y la sustancia de referencia química (std) que absorben en la longitud de onda máxima correspondiente al principio activo.
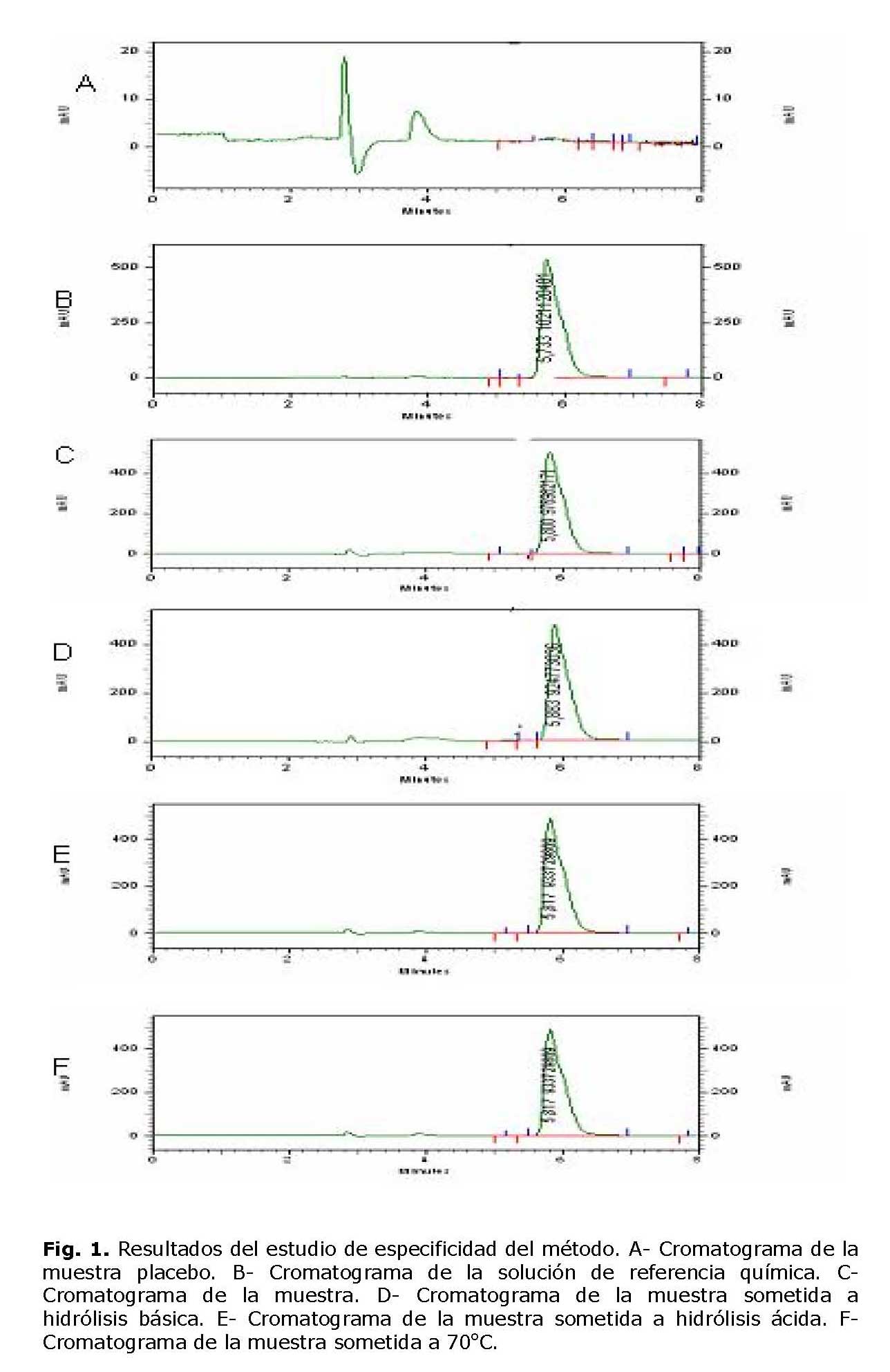
La figura 1 muestra los resultados del estudio de especificidad del método cromatográfico. Como se observa en el cromatograma correspondiente a la muestra placebo (A) no se obtuvo ninguna señal en la zona de interés (tr 5.8 minutos), al ser comparado con la señal obtenida para la solución estándar de referencia (B) y de la muestra de Paracetamol gotas orales(C), lo cual indica que los excipientes no interfieren en la determinación del principio activo. En las muestras sometidas a condiciones drásticas (D, E, F) se evidenció una ligera disminución de los picos correspondientes al Paracetamol, no se observó aparición de picos secundarios, atribuibles a productos de degradación del principio activo.
En la tabla 1, se reportan los resultados de los estudios de la linealidad del sistema, para el método cromatográfico y espectrofotométrico, el coeficiente de regresión lineal fue de 0,9998 y 0,9998, respectivamente, y los coeficientes de variación del factor de respuesta resultaron 4,13 % y 2,56 % en cada caso.
Tabla 1. Resultados del estudio de linealidad y exactitud
| Resultados del estudio de linealidad | |||||||
| Parámetros | Cromatográfico | Espectrofotométrico | Límites | ||||
| Ecuación de la recta | Y = 0,3516 X + 7,77 | Y = 0,07 x + 0,03 | y = bx + a | ||||
| Coeficiente de regresión lineal | 0,9998 | 0,9998 | ≥ 0,9990 | ||||
| Desviación estándar relativa de la pendiente | 0,99 | 0,91 | ≤ 2 % | ||||
| Coeficiente de variación del factor de respuesta | 4,13 | 2,56 | ≤ 5 % | ||||
| Resultados del estudio de exactitud | |||||||
| Niveles | Cromatográfico | Espectrofotométrico | Límites | ||||
| Recuperación (%) | Resultados | Recuperación (%) | Resultados | ||||
| 80 % | 100,06 100,04 99.98 | R media = 100,00 % t calc. = 1,740 t tab. = 2,306 G calc. = 0,420 G tab. = 0,767 | 99,00 100,02 99,62 | R media = 99,75 % t calc. = 1,680 t tab. = 2,306 G calc. = 0,482 G tab. = 0,767 | 98,0 – 102,0 % t exp. ≤ t tab. G exp. ≤ G tab. | ||
| 100 % | 100,03 99,99 99,95 | 99,8 99,5 100,1 | |||||
| 120 % | 100,05 99,98 100,00 | 99,67 100,17 99,83 | |||||
Leyenda: R, porcentaje de recuperación; t, distribución t students; G, coeficiente de la prueba de Cochran; Calc., calculada; tab., tabulada.
En la tabla 2, aparecen reportados los resultados de los estudios de exactitud, para el método cromatográfico y espectrofotométrico. Las recuperaciones medias fueron de 100,00 % y 99,75 %, respectivamente, y los valores de t calculadas (1,74 y 1,68) y de G calculada (0,420 y 0,484) fueron menores que los valores tabulados, para un 95 % de confianza, t tabulada (2,306) y G tabulada (0,797).
Tabla 2: Resultados del estudio de la precisión intermedia del método analítico (CLAR) y (UV)
| Resultados de precisión por CLAR | ||||||||||||||
| Niveles | Analista 2 (%) | |||||||||||||
| Primer día | Segundo día | Tercer día | Primer día | Segundo día | Tercer día | |||||||||
| 80 % | 80,12 | 80,03 | 80,01 | 80,12 | 80,06 | 80,06 | ||||||||
| 80,06 | 80,06 | 79,98 | 80,06 | 80,09 | 80,00 | |||||||||
| 80,05 | 80,08 | 80,03 | 79,98 | 80,02 | 79,97 | |||||||||
| 100 % | 99,97 | 100,03 | 99,98 | 100,09 | 99,97 | 100,02 | ||||||||
| 99,99 | 100,05 | 99,96 | 100,07 | 99,95 | 100,04 | |||||||||
| 100,06 | 99,98 | 100,04 | 99,98 | 100,03 | 99,97 | |||||||||
| 120 % | 119,98 | 119,89 | 120,06 | 119,97 | 120,04 | 119,96 | ||||||||
| 119,89 | 119,96 | 120,03 | 120,04 | 119,98 | 120,06 | |||||||||
| 120,03 | 120,02 | 119,97 | 120,06 | 119,95 | 119,94 | |||||||||
| Análisis Estadístico | ||||||||||||||
| Prueba de significación de Fisher por analistas ( F tab ( 8/8; 0.05) = 3,44) | Prueba de significación de Fisher por día ( F tab (5/5; 0.05) = 5,05 ) | Límites | ||||||||||||
| Niveles | Días 1/2 | Días 2/3 | Días 1/3 | F exp ≤ F tab | ||||||||||
| 80 | 1,56 | 80 | 3,62 | 1,47 | 2,47 | |||||||||
| 100 | 1,59 | 100 | 1,67 | 1,25 | 2,12 | |||||||||
| 120 | 0,65 | 120 | 1,35 | 1,01 | 1,37 | |||||||||
| Prueba de significación de student por analistas (t tab (16; 0.05) =2,12) | Prueba de significación de student por días ( t tab ( 10; 0,05) = 2,22) | Límites | ||||||||||||
| Niveles | Días 1/2 | Días 2/3 | Días 1/3 | |||||||||||
| 80 | 0,82 | 80 | 0,34 | 2,07 | 2,15 | t exp ≤ t tab | ||||||||
| 100 | 0,89 | 100 | 0,92 | 0,18 | 0,96 | |||||||||
| 120 | 1,19 | 120 | 0,65 | 0,97 | 0,25 | |||||||||
| Coeficiente de Variación | ||||||||||||||
| Niveles | Analista 1 | Analista 2 | Límites | |||||||||||
| 80 | 0,05 | 0,06 | CV ≤ 2,0 % | |||||||||||
| 100 | 0,04 | 0,05 | ||||||||||||
| 120 | 0,05 | 0,04 | ||||||||||||
| Resultados de precisión por UV | ||||||||||||||
| Analista 1 (%) | Analista 2 (%) | |||||||||||||
| Día 1 | Día 2 | Día 1 | Día 2 | |||||||||||
| 99,4 | 99,1 | 100,0 | 100,1 | |||||||||||
| 99,0 | 99,3 | 99,5 | 99,2 | |||||||||||
| 99,6 | 100,3 | 99,4 | 99,4 | |||||||||||
| 100,0 | 99,4 | 99,9 | 99,5 | |||||||||||
| 99,8 | 99,8 | 100,2 | 99,7 | |||||||||||
| t cal 1 ═ 0,15 ; t cal 2 ═ 0,25 t tab(5%) ═ 2,07 F cal 1 ═ 1,98; F cal 2 ═ 1,16 Ftab (5%) ═ 2,97 | ||||||||||||||
Leyenda: C.V. Coeficiente de variación; X, valor medio; CLAR, cromatografía líquida de alta resolución; UV, espectrofotometría ultravioleta
Los resultados de los estudio de precisión de los métodos desarrollados aparecen reportados en las tablas 3 y 4, En los estudios de repetibilidad realizados, para el método cromatográfico y espectrofotométrico, las medias obtenidas fueron de 99,98 % y 99,60 %, y los coeficientes de variación fueron de 0,14 % y 0,35 %, respectivamente, mientras que los valores de F calculadas y los valores de t calculadas fueron menores que los valores tabulados, para un 95 % de confianza, para cada uno de los niveles estudiados.
Tabla 4: Estudio de vida de estante
| Parámetros | Tiempo (meses) | Lotes | ||
| 9001 | 9002 | 9003 | ||
| Características | 0 | Responde | Responde | Responde |
| 3 | Responde | Responde | Responde | |
| 6 | Responde | Responde | Responde | |
| 9 | Responde | Responde | Responde | |
| 12 | Responde | Responde | Responde | |
| pH | 0 | 5,62 ± 0,823 | 5,64 ± 0,848 | 5,61 ± 0,621 |
| 3 | 5,90 ± 0,742 | 5,85 ± 0,706 | 5,85 ± 0,886 | |
| 6 | 5,90 ± 0,589 | 5,93 ± 0,719 | 5,93 ± 0,557 | |
| 9 | 5,87 ± 0,724 | 5,84 ± 0,646 | 5,88 ± 0,639 | |
| 12 | 5,94 ± 0,567 | 5,92 ± 0,329 | 5,90 ± 0,337 | |
| Valoración | 0 | 99,3 ± 0,93 | 99,1 ± 0,42 | 98,6 ± 0,83 |
| 3 | 98,7 ± 0,58 | 98,5 ± 0,73 | 97,8 ± 0,52 | |
| 6 | 98,1 ± 0,62 | 98,2 ± 1,07 | 96,9 ± 0,77 | |
| 9 | 98,0 ± 0,47 | 98,0 ± 0,45 | 96 6 ± 0,39 | |
| 12 | 97,6 ± 0,53 | 97,8 ± 0,39 | 96,1 ± 0,51 | |
| Productos de degradación | 0 | Inapreciables | Inapreciables | Inapreciables |
| 3 | Inapreciables | Inapreciables | Inapreciables | |
| 6 | Inapreciables | Inapreciables | Inapreciables | |
| 9 | Inapreciables | Inapreciables | Inapreciables | |
| 12 | Inapreciables | Inapreciables | Inapreciables | |
| Conteo microbiológico | 0 | CB: < 10 UFC/mL CH: < 10 UFC/mL | CB: < 10 UFC/mL CH: < 10 UFC/mL | CB: < 10 UFC/mL CH: < 10 UFC/mL |
| 12 | CB: 10 UFC/mL CH: < 10 UFC/mL | CB: 10 UFC/mL CH: < 10 UFC/mL | CB: 10 UFC/mL CH: < 10 UFC/mL | |
Leyenda: CB: conteo de bacterias, CH: conteo de hongos, UFC: unidades formadoras de colonia
Estudio de estabilidad
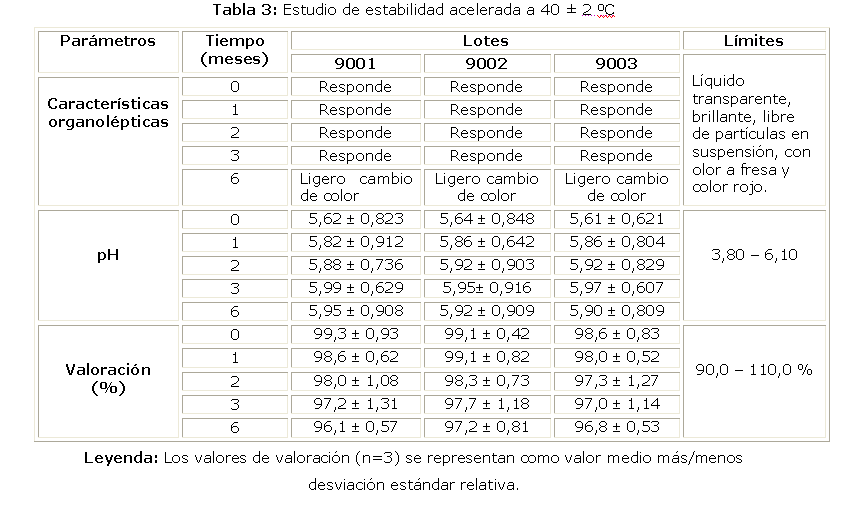
Los resultados del estudio de estabilidad acelerada a 40 ± 2ºC, correspondiente a los lotes 9001, 9002 y 9003, y se demuestra que el producto terminado no presentó cambios significativos en pH, valoración, si se evidenció un ligero cambio de color en la formulación a los 6 meses de transcurrido el estudio.
En los resultados del estudio de vida de estante hasta 12 meses se observó que el producto terminado cumplió con las especificaciones de calidad durante el estudio.
En los resultados del estudio de frasco en uso, se demuestró que el producto se mantuvo estable durante los 30 días estudiados.
DISCUSIÓN
Validación de los métodos analíticos
Los valores obtenidos en el estudio de especificidad del método espectrofotométrico demostraron que las sustancias auxiliares no interfieren a la longitud de onda correspondiente al máximo de absorción del principio activo (245 nm), ya que los valores de absorbancia del placebo corresponden al 1 % con relación a los valores de Paracetamol en las muestras, por tanto, es adecuado para el control de la calidad del producto terminado.8-9
No se observó interferencias de los productos de degradación en la determinación del principio activo. Se apreció una ligera disminución del pico del Paracetamol, en los medios de degradación utilizados, sin aparición de picos secundarios. No se evidenció interferencia de los excipientes utilizados en la formulación, en el intervalo de elución del principio activo, demostrándose la especificidad del método cromatográfico tanto para el control de la calidad, como para realizar el estudio de estabilidad de las gotas orales.8-9
Los resultados del estudio de linealidad (tabla 1) probaron que los métodos cumple con los parámetros establecidos, el coeficiente de correlación de la recta de regresión es mayor que 0,999 para ambos métodos, los factores respuestas son semejantes entre sí, similares al valor de la pendiente de la recta de regresión y el coeficiente de variación de éstos menores que 5%. Al aplicarse la prueba de proporcionalidad este resultó no significativo (SBrel< 2,0%) y el intervalo de confianza del intercepto incluye el valor cero en cada método.
El estudio de la exactitud (tabla 2) demostró que los métodos cumplen con los requisitos establecidos para este parámetro. Los valores de recuperación media se encuentran dentro de los límites establecidos (98 102 %), con un coeficiente de variación dentro de los límites establecidos. El valor de t experimental fue menor que el tabulado para un 95 % de confianza y grados de libertad,8 lo que señala que el método es exacto, con una certeza del 95 %, para la cuantificación del principio activo y que no existieron desviaciones por exceso o por defecto. Tampoco el factor concentración influyó en la variabilidad de los resultados pues los valores de G exp fueron menores que el tabulado para una probabilidad de 0,05, k=3 y n=38.
Los ensayos de precisión (tablas 3 y 4) indicaron que no existían diferencias significativas entre las precisiones alcanzadas por ambos analistas, en días diferentes, ni en las medias obtenidas por ellos. Los valores que se obtienen en el estudio de precisión intermedia, de las pruebas de Fischer y t - Student, demuestran que no existen diferencias significativas entre las precisiones alcanzadas por los analistas en diferentes días para una probabilidad de 0,05, ya que el valor de F calculada es menor que la F tabulada. Al realizar la prueba de t- Student el valor calculado resultó menor que el tabulado, para una probabilidad de 0,05, lo cual demostró que no existían diferencias significativas entre las medias alcanzadas, con un nivel de significación de un 5%.8
El método por espectrofometría ultravioleta puede emplearse en el control de calidad del producto terminado al igual que el método cromatográfico, siendo el de elección por su sencillez y rapidez el espectrofotométrico y el método por cromatografía líquida de alta resolución para el estudio de estabilidad del producto por su elevada especificidad.
Estudio de estabilidad
El conjunto de los resultados del estudio de estabilidad acelerada a 40 ± 2ºC, demostraron la estabilidad del producto. Durante el período evaluado, los 3 lotes analizados mantuvieron las características organolépticas, el pH y la concentración del principio activo. Se demostró la estabilidad térmica del fármaco ya que después de transcurridos 6 meses se mantuvo la concentración conforme con los límites establecidos en las especificaciones de calidad del producto terminado.4-8 A los 6 meses se observó un color rojo con una ligera oscuridad, este cambio no resultó atribuible a la degradación del fármaco puesto que este mantuvo su valoración dentro de los límites establecidos, este comportamiento concuerda con lo esperado puesto que durante la validación de método, en la evaluación de la especificidad, se observó que el medicamento no se degradó tras su exposición a 70 C; la disminución del principio activo en el ensayo de valoración resultó menor del 5 %, por lo que no se consideró un cambio significativo.
De los resultados obtenidos, se infiere que el producto mantuvo los parámetros que determinan su calidad, tanto en su etapa inicial como transcurridos 12 meses. Además, no se observaron cambios en los aspectos organolépticos, los valores obtenidos para el pH se encontraron dentro de los límites establecidos en las especificaciones de calidad del producto terminado, y en la valoración durante el tiempo de almacenamiento estudiado no se observó una disminución del principio activo superior al 5 %. Los resultados del conteo microbiológico realizado al inicio y al final de estudio demostraron la estabilidad microbiológica del producto terminado.
Los resultados del estudio de frasco en uso de las gotas orales, se encuentran dentro de los límites establecidos para el producto terminado, lo que demuestra que el medicamento se mantiene estable durante el uso de este por los pacientes que lo necesitan en su tratamiento, cuando se almacena a temperatura ambiente. No existieron diferencias significativas (p < 0,05) entre las medias obtenidas en el tiempo inicial y el tiempo final de estudio.
Los resultados de los estudios de estabilidad acelerada y de vida de estante, , demostraron la estabilidad física, química y microbiológica del producto por espacio de 12 meses a temperatura inferior a 30 ºC, ya que todas las variables analizadas se encontraron dentro de los límites establecidos en las especificaciones de calidad para las gotas orales de Paracetamol 100 mg/mL.
Por todo lo anterior, se puede concluir que los métodos analíticos por espectrofotometría UV y cromatografía líquida de alta resolución, son válidos para el control de la calidad y estudio de estabilidad de las gotas orales de Paracetamol 100 mg/mL, ya que resultaron lineales, precisos, exactos y específicos.
Se demostró la estabilidad física, química y microbiológica del producto por espacio de 12 meses a temperatura inferior a 30 ºC, envasados en frascos de vidrio ámbar por 15 mL, boca 18 mm, calidad hidrolítica III. Además se evidenció que el producto es estable durante 30 días después de abierto el frasco.
REFERENCIAS BIBLIOGRÁFICAS
1. Goodman A, Gilman, A. Las bases farmacológicas de la terapéutica. Tomo II. 3ra ed. La Habana: Editorial Científico Técnica.1994. pp: 285 -287.
2. PDR. Physicians´ Desk Reference. 57 edición. Inc at montuale, New York, Estados Unidos; 2003, p. 1025
3. Quattrocchi O.A. & Laba R.F. «Introducción al HPLC» en "Aplicación y práctica". Ed. Artes Gráficas Farro S.A, Buenos Aires. Pág. 106-122, 284, 302-328. 1992.
4. Dierksneier, G. "Métodos Cromatográficos". Ed. Científico - Técnica. La Habana, Cuba. págs. 1- 4, 256 - 412. 2005
5. Resolución 49/04. Regulación 37/04 Buenas Prácticas de Laboratorio para el Control de Medicamentos. La Habana: Centro Estatal para el Control de Medicamentos (CECMED). Fecha de acceso: Agosto del 2004. Disponible en http://www.cecmed.sld.cu/Pages/AmbReg-3.htm
6. Resolución 34. Regulación 06/01. Requerimientos de los estudios de estabilidad para el registro de productos farmacéuticos nuevos o conocidos. La Habana: Centro Estatal para el Control de Medicamentos (CECMED). Fecha de acceso: 28 de septiembre del 2000. Disponible en http://www.cecmed.sld.cu/Pages/Reg_LicProd.htm
7. World Health Organization. Stability of Drug Dosage Forms. Expert Committee on Specification for Pharmaceutical Preparations, Thirty first Report. WHO Thecnical Report. Geneva: WHO; 1996.
8. Resolución 18. Regulación 41/07. Validación de Métodos de analíticos. La Habana: Centro Estatal para el Control de Medicamentos (CECMED). Fecha de acceso: 26 de marzo del 2007. Disponible en http://www.cecmed.sld.cu/Pages/Reg_EvalEL.htm
9. Farmacopea de los Estados Unidos de América (USP 31 NF 26). Twinbrook Parkway, Rockville, MD, Estados Unidos de América: Ed. The United States Pharmacopeial Convention. 2010: 1388.
Recibido: 24 de septiembre de 2012.
Aprobado: 4 de noviembre de 2012.
MSc. Caridad M. García Peña
Centro de Investigación y Desarrollo de Medicamentos, Ave 26 # 1605, La Habana, Cuba
Teléfono 8810882, 8811944, 8811424, Fax (537) 335556, correo electrónico caridadgp@infomed.sld.cu

{kind=link}
{kind=link}