










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El trastorno del espectro autista (TEA) es un conjunto de complejas alteraciones del neurodesarrollo que se caracteriza por dificultades en la interacción social, así como conductas repetitivas y restringidas. Su diagnóstico es clínico y tiene un origen multifactorial.1,2,3,4,5) Aproximadamente uno de cada 160 niños tiene un TEA.6
Por su parte, la epilepsia es una enfermedad cerebral definida por cualquiera de las siguientes circunstancias: 1) al menos dos crisis no provocadas (o reflejas) con más de 24 h de separación, 2) una crisis no provocada (o refleja) y una probabilidad de presentar nuevas crisis durante los 10 años siguientes similar al riesgo general de recurrencia (al menos, 60 %) tras la aparición de dos crisis no provocadas, 3) diagnóstico de un síndrome de epilepsia.7
En 2017 la Liga Internacional contra la Epilepsia publicó una clasificación sobre los tipos de crisis epilépticas, en la que las dividía en tres grupos de acuerdo a su inicio (motor o no motor): focal, generalizado o desconocido.8
Según reportes de la Organización Mundial de Salud, más de 50 millones de personas en el mundo padecen de epilepsia, lo que la convierte en una de las enfermedades neurológicas más comunes; cerca de 80 % de los pacientes viven en países de bajos ingresos. Afecta al 1-2 % de la población.9,10
La epilepsia es más común entre individuos con TEA que en la población general; esta comorbilidad es frecuente y su diagnóstico es todo un reto.11,12,13,14,15) Entre 8-30 % de los pacientes pediátricos con TEA también padecen epilepsia.16
Se desconoce la existencia de publicaciones realizadas en Cuba que relacionen el TEA con la epilepsia en edades pediátricas; tampoco es profusa la producción científica al respecto en América Latina.
Ello motivó la realización de esta investigación, cuyo objetivo fue caracterizar la comorbilidad del TEA y la epilepsia en pacientes pediátricos.
Métodos
Estudio observacional, descriptivo, de corte transversal, realizado entre el 1 de enero de 2020 y el 1 de noviembre de 2021 en el Centro Internacional de Restauración Neurológica (CIREN), institución médica en la que se atendieron los pacientes de esta investigación.
La población estuvo constituida por 23 individuos y fue estudiada en su totalidad. Los criterios de inclusión fueron: todos los pacientes en edad pediátrica (≤ 19 años) con diagnóstico de TEA [según los criterios de la quinta edición del Manual Diagnóstico y Estadístico de los Trastornos Mentales (DSM-5) 17] y epilepsia atendidos en consulta externa de neuropediatría del CIREN en el citado período, cuyos tutores legales dieran el consentimiento escrito a que se les incluyera. Se excluyeron aquellos pacientes con historia clínica (HC) incompleta.
Se estudiaron las variables: sexo (masculino o femenino), edad del paciente en la primera consulta (en años cumplidos: 0-9, 10-14, 15-19), edad del paciente al momento del diagnóstico del TEA (en años cumplidos: 0-9, 10-14, 15-19), edad del paciente en la primera crisis epiléptica (en años cumplidos: 0-9, 10-14, 15-19), edad gestacional del paciente al momento de su nacimiento (menos de 32 semanas, de 32 a 37 semanas, de 38 a 42 semanas o más de 42 semanas), trastorno de déficit intelectual (sí o no, de acuerdo a su diagnóstico), factores de riesgo (infecciones durante el embarazo, consumo de alcohol de la madre durante el embarazo, exposición a la nicotina durante el embarazo, antecedentes patológicos maternos de epilepsia o ninguno), tipo de crisis epiléptica (focal, generalizada o de inicio desconocido), tipo de TEA (congénito o regresivo, según su diagnóstico), grado del TEA según puntaje (ligero o moderado: 30-36 puntos; profundo: más de 36 puntos) obtenido en la escala de autismo infantil18 (CARS, del inglés Child Autism Rating Scale) y causa del TEA [síndrome de Rett, síndrome de Down, síndrome de West, síndrome de Angelman, lesión estática en el sistema nervioso central sin otras complicaciones (LESNC-SOC), síndrome de Lennox-Gastaut o idiopático].
Los datos se recolectaron de las HC individuales en planillas diseñadas con tal efecto, y luego se exportaron a hojas de trabajo de Microsoft Excel 2016 para Windows.
Se realizó un análisis estadístico descriptivo con frecuencias absolutas y porcentuales. Se determinaron medidas de tendencia central (moda y media) y de dispersión (desviación estándar). Se empleó el programa estadístico IBM SPSS 26 para Windows.
Se respetaron los principios bioéticos clásicos de autonomía, beneficencia, no maleficencia y justicia. Se cumplió con lo refrendado en la Declaración de Helsinki19 sobre investigaciones médicas en seres humanos. Los tutores legales de los individuos que participaron en el estudio expresaron su autorización mediante la firma de un consentimiento informado. Se protegió la confidencialidad de los datos de los sujetos estudiados, y de sus representantes legales.
Resultados
Predominaron los pacientes del sexo femenino (n= 14; 60,9 %). La totalidad de los individuos
(n= 23; 100 %) acudió a la primera consulta, fue diagnosticado con TEA y sufrió la primera crisis epiléptica con 9 años de edad o menos; estos eventos se produjeron, respectivamente, a las edades medias de 3,57±2,16 años, 2,35±1,49 años y 3,1±2,03 años. El 78,3 % (n= 18) nació entre las 38 y 42 semanas de gestación, y el 65,2 % (n= 15) se diagnosticó con trastorno de déficit intelectual. Lo más común fue la inexistencia de factores de riesgo (n= 14; 60,9 %); pero, cuando existieron, los de mayor representación fueron las infecciones y la exposición a la nicotina, ambos presentes en 3 casos diferentes, lo cual representó 13 % para cada uno (Tabla 1).
Tabla 1 -Distribución de los pacientes según variables estudiadas
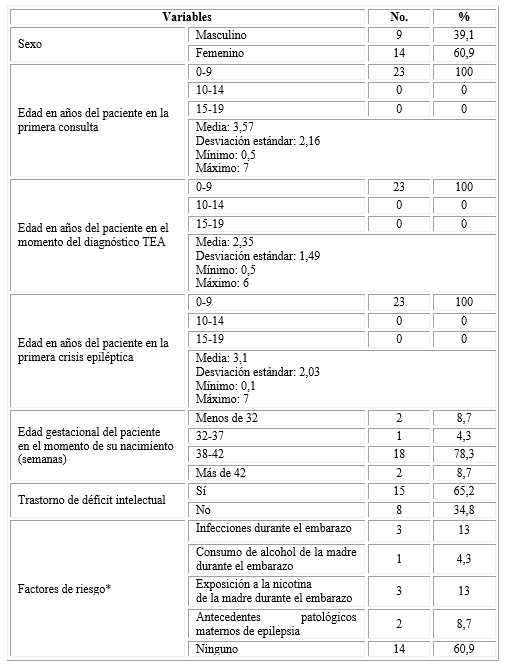
Tamaño de la población: 23.
*En ningún caso se presentó más de un factor de riesgo a la vez.
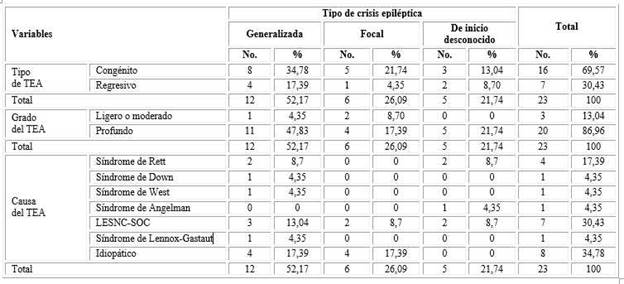
Las crisis epilépticas generalizadas fueron las más frecuentes (n= 12; 52,17 %).
El TEA congénito se presentó más comúnmente entre los pacientes estudiados (n= 16; 69,57 %), fundamentalmente en aquellos con crisis generalizadas (n= 8; 34,78 %). La mayoría de los individuos presentó un TEA profundo (n= 20; 86,96 %), igualmente con predominio de la presencia de crisis generalizadas (n= 11; 47,83 %).
La causa más frecuente de TEA fue la LESNC-SOC (n= 7; 30,43 %), solo superada en número por el origen idiopático (n= 8; 64,78) (Tabla 2).
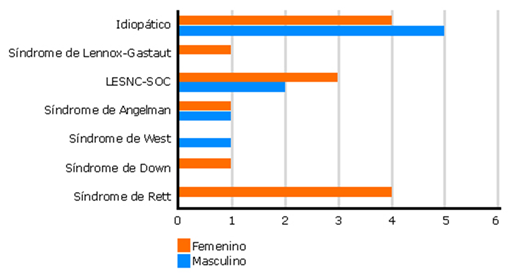
El síndrome de West fue la única causa que no tuvo representación femenina y solo se presentó en un paciente. Por otra parte, los síndromes de Lennox-Gastaut (n= 3), Down (n= 1) y Rett (n= 4) solo se presentaron en pacientes femeninas. El origen idiopático fue el más frecuente (n= 9), fundamentalmente en el sexo masculino (n= 5) (Fig. 1).
En más de la mitad de la población (n= 13; 56,5 %), la primera crisis epiléptica antecedió en el tiempo al diagnóstico del TEA (Fig. 2).

Discusión
Los resultados en torno al sexo de los pacientes coinciden con los estudios de otros investigadores20,21 en los que también se evidenció una superioridad del sexo femenino con coexistencia de epilepsia y TEA; sin embargo, estudios realizados en otros países documentan una mayor incidencia en el sexo masculino para ambas afecciones por separado.10,22,23
En este estudio, la edad de la primera consulta tuvo un rango de 6,5 años. Otros autores encuentran para esta misma variable un rango de 17 años.24 El resultado en esta investigación, considerablemente menor, es comprensible si se tiene en cuenta que ninguno de los sujetos que se incluyeron en esta investigación rebasaron los 9 años de edad.
Algunos investigadores25 en su estudio observan que 42 % de los pacientes con TEA y epilepsia experimentaron la primera crisis epiléptica entre los 10 y 19 años, rango de edad al que no perteneció ningún sujeto de este estudio.
El predominio notable (78,3 %) de los pacientes nacidos con edad gestacional entre 38 y 42 semanas posee semejanza con el obtenido en un estudio poblacional,26 en el cual estos pacientes representaron 86,1 %. Pudiera esto sugerir que la comorbilidad entre TEA y epilepsia no está vinculada al nacimiento pre o postérmino del paciente.
Los resultados obtenidos en la presente investigación relacionados con el trastorno de déficit intelectual y TEA congénito asociado es similar a otra27) que plantea que 40 % de sus pacientes exhibían este trastorno.
El 20 % de las madres, en un estudio en pacientes con TEA controlados en un hospital de niños,28 consumieron sustancias tóxicas en el embarazo, dato casi coincidente con el de este estudio: 17,3 %.
Tanto en esta investigación, como en otras,26,29 predominaron las crisis epilépticas generalizadas, con una frecuencia relativa porcentual cercana a 60 %. Es esto congruente con el hecho de que las crisis epilépticas generalizadas son las de mayor frecuencia de presentación.30
Un estudio ecuatoriano25 y el aquí presentado registran una mayor frecuencia de TEA profundo entre este tipo de pacientes.
Es interesante cómo el síndrome de Rett, pese a su considerable representación en la población estudiada, solo se presentó en féminas, lo cual puede ser explicado por su prevalencia mundial considerablemente mayor para el sexo femenino que para el masculino.31
Este estudio posee la limitación de que la recolección de los datos fue de HC de un centro de consulta externa, donde se espera que pacientes con ambas enfermedades asistan por descompensación con menor frecuencia en comparación con un centro hospitalario.
No obstante, esta investigación tiene la virtud y originalidad de no poseer ningún antecedente, por lo menos publicado en Cuba, por lo cual podrá convertirse en punto de partida para la ejecución de futuros estudios de esta comorbilidad, y la discusión de sus resultados, tanto en el ámbito nacional como regional.
Se concluye que, en la población estudiada, la comorbilidad entre epilepsia y TEA se produce fundamentalmente en el sexo femenino, antes de la primera década de la vida, con coexistencia frecuente de un trastorno de déficit intelectual y la ausencia de factores de riesgo, en el contexto de crisis epilépticas generalizadas y un TEA congénito, profundo, idiopático y con diagnóstico posterior al de la epilepsia.

