










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Las malformaciones congénitas del estómago tienen poca frecuencia; dentro de ellas las más reportadas: las duplicidades gástricas, los islotes de páncreas ectópico, los diviertículos gástricos y la atresia pilórica.
La atresia pilórica es una afección rara, registrada por primera vez en 1749 por Calder. Esta afección tiene una incidencia de 1 x 100 000 nacidos vivos y según su clasificación anatómica puede ser: tipo 1, membrana pilórica, tipo 2, tejido pilórico remplazado por tejido sólido, y tipo 3, atresia pilórica con discontinuidad gastro-duodenal. Además, esta malformación se presenta de forma aislada o asociada con otras anomalías en el 40-50 % de los casos, de ellas la más frecuente es la epidermolisis bullosa, enfermedad también poco frecuente que se presenta en 1 x 300 000 nacidos vivos.1,2,3) La epidermólisis bullosa abarca un grupo de trastornos hereditarios que presentan una amplia variabilidad fenotípica debido a que la herencia de cada uno de ellos está ligada a la mutación de diferentes genes que codifican diferentes proteínas de la unión epidermodérmica. Se trata de una enfermedad de piel y mucosas que se caracteriza por fragilidad cutánea leve a severa y ampollas o erosiones, luego de traumas mínimos. Se conocen tres formas clínicas, dependiendo del lugar donde aparezca la alteración estructural y del nivel epidérmico: epidermólisis ampollosa simple, epidermólisis ampollosa juntural, y epidermólisis ampollosa distrófica.2,4) La asociación entre atresia pilórica y epidermolisis bullosa se denomina síndrome de Carmi.1) Swinburne y Kohler publicaron por primera vez esta asociación en 1968, y en 1982, Carmi describió 2 casos de aplasia cutis congénita, uno de ellos asociado con atresia pilórica. un año después apareció en la literatura el síndrome de Carmi para designar la aparición conjunta de epidermólisis ampollosa de la unión con atresia gastrointestinal congénita, con mayor frecuencia atresia pilórica y aplasia cutis congénita. Este síndrome, por histopatología, se debe a ausencia de la integrina α6β4 integrin y alteración en la laminilla-322.5,6
El propósito de esta presentación fue informar sobre la evolución de una paciente tratada por atresia pilórica que tenía, además, una epidermolisis bullosa.
Presentación del caso
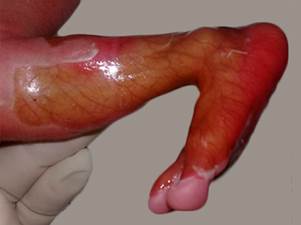
Recién nacida con antecedentes prenatales de polihidramnios, infección urinaria en el segundo trimestre del embarazo. Con parto eutócico a las 30,4 semanas, sepsis ovular materna, peso al nacer 1430 g, Apgar 7-8, que se ventiló desde la sala de partos y se reportó de crítica desde su admisión en sala de neonatología. Presentó múltiples lesiones en piel, ampollosas, en todo el cuerpo, con aplasia cutis en pierna izquierda (fig. 1).
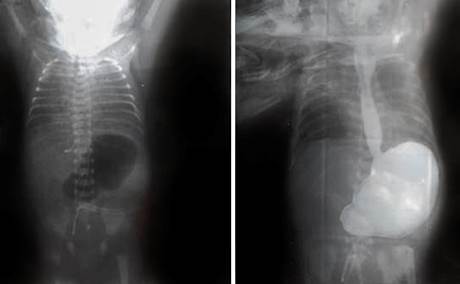
Se retiró del ventilador a las 48 h y se comenzó la alimentación enteral mínima, que no toleraba. En la radiografía de abdomen simple y contrastado se apreció ausencia de gas en el tracto gastrointestinal con una cámara gástrica dilatada y no paso de contraste al píloro.
Se interconsultó con cirugía y se planteó una atresia pilórica (fig. 2).
La paciente estaba con estabilidad hemodinámica y se descartaron otras anomalías que pudiesen estar asociadas. Defecó (lo que excluye otras malformaciones intestinales) y se decidió tratamiento quirúrgico al cuarto día de nacida con todas las medidas preoperatorias requeridas. En el acto quirúrgico se constató atresia pilórica tipo 2: sustitución del tejido pilórico por tejido fibroso. Se realizó una gastroduodenostomía (fig. 3).
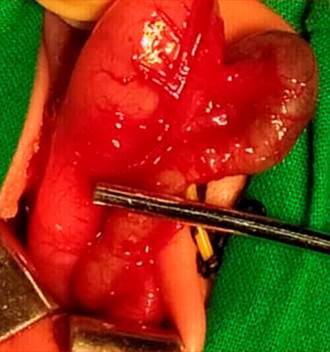
En su evolución se incrementaron por día las lesiones en piel, ya sea de forma espontánea y principalmente en áreas de traumatismos (lugares de apoyo, herida quirúrgica, lugares de vendajes, venipunturas). Tuvo reapertura del ductus arterioso, trastornos hidroelectrolíticosy hemidinámicos, que provocaron el fallecimiento a los 14 días de nacida.
Discusión
La presencia de polihidramnios se encuentra en la atresia pilórica como en el caso presentado, aunque no tuvo un diagnóstico prenatal de esta afección. Si se sospecha por ecografía prenatal que el paciente tiene una atresia pilórica, estómago fetal distendido y ausencia de intestino dilatado distalmente, debe estudiarse la posibilidad de que tenga asociada una epidermolisis bullosa. Se detecta en estos casos el signo ecográfico de “copos de nieve”, producto de las escamas de piel fetal en el líquido amniótico.3
La atresia pilórica, generalmente se presenta con vómitos biliosos después del nacimiento. La paciente en este trabajo, tuvo el antecedente de prematuridad, bajo peso, necesidad de ventilación mecánica al nacer, y , presentó ,una intolerancia a la alimentación enteral mínima, aumento del contenido drenado por la sonda nasogástrica, con sospecha de atresia pilórica al tercer día de nacida.2
Aunque este caso no tuvo otras malformaciones asociadas, en la literatura se publica las asociaciones de atresia pilórica con otras anomalías como: atresia duodenal, atresias múltiples de colon, atresia esofágica, anomalías renales o ureterales. No tuvo otras malformaciones que pueden acompañar a la epidermolisis bullosa.1,2
Esta paciente presentó una variedad anatómica tipo 2, que aparece en 34% de los casos; el tipo 1 es el más frecuente (membrana pilórica) en el 57 % y la menos frecuente el tipo 3 (atresia pilórica con discontinuidad gastro-duodenal). en 9 % de los casos.2,3
La epidermolisis bullosa se clasifica en cuatro grandes grupos. De acuerdo con el lugar dentro de la dermis de formación de la ampolla, puede ser: simple, de la unión, distrófica y el síndrome de Kindler. Esta enfermedad, según la distribución de las lesiones, se clasifica en: localizada o generalizada y por la identificación de la proteína afectada mediante inmunoflorescencia. La paciente en estudio tenía una epidermolisis bullosa de la unión (junction) y dentro de ella, la variedad de asociación con atresia pilórica y generalizada.5
La atresia pilórica, cuando se asocia con otras anomalías, tiene una alta mortalidad, y cuando presenta un síndrome de Carmi, o sea, se asocia con epidermolisis bullosa, la mortalidad es mucho mayor, sobre todo por la presencia de manifestaciones sistémicas de desbalances hidroelectrolíticos, septicemia, malnutrición y morbilidades respiratorias. Por todo esto resulta controversial la indicación quirúrgica en estos casos.1,5,7
En ausencia de epidermólisis bullosa y de otras anomalías asociadas, la atresia de píloro representa una malformación corregible quirúrgicamente y con un buen pronóstico.1
El asesoramiento genético es fundamental para familias con atresia pilórica con epidermolisis bullosa como el caso presentado. Los padres son portadores; se trata de un trastorno autosómico recesivo, y existe una probabilidad de cada cuatro embarazos de presentar la enfermedad.8
Se concluye que la atresia pilórica es una afección muy rara, que debe tenerse en cuenta en recién nacidos con epidermolisis bullosa por la frecuente asociación entre estas dos afecciones; además, cuando existen antecedentes de polihidramnios y no tolerancia a la alimentación enteral. Los pacientes con la asociación atresia pilórica y epidermolisis bullosa generalmente tienen una evolución desfavorable.

