










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La insuficiencia adrenal (IA) es una condición clínica causada por una secreción inadecuada de las hormonas esteroideas, principalmente de los glucocorticoides. Se puede clasificar en insuficiencia adrenal primaria (adrenal), secundaria (hipófisis) o terciaria (hipotálamo).1,2 En la edad pediátrica se presenta la primaria como principal causa de IA, que se manifiesta esencialmente en neonatos y lactantes; originada, frecuentemente, por condiciones genéticas como hiperplasia adrenal congénita, adrenoleucodistrofia o hipoplasia adrenal congénita, entre otras.1,3,4
La secundaria se asociada comúnmente a tumores hipofisarios o adyacentes a la zona, con deficiencia de la hormona adrenocorticotropa (ACTH) por el tumor o como secuelas del tratamiento. Es más común hallarla en combinación con el déficit de otras hormonas hipofisarias o presentarse aislada. Lo más frecuente consiste en que se produzca por mutaciones recesivas en el gen TBX19 (anteriormente denominado TPIT, factor de transcripción T-BOX), un factor de transcripción encargado de la diferenciación terminal de las células hipofisarias que expresan proopiomelanocortina (POMC).3,5,6 Por otro lado, la terciaria muestra como principal causa el tratamiento con corticoides exógenos de uso prolongado en enfermedades como el asma, síndrome nefrótico, oncológicas, entre otras, que suprimen el eje hipotálamo-hipofisario-adrenal y, al retirarlos abruptamente, desarrollan la IA.5
En la población pediátrica la incidencia de la IA no está bien descrita. Se conocen datos casi exclusivamente de la hiperplasia adrenal congénita, que se presenta en 1/10 000-18 000 recién nacidos.7 Con respecto a la crisis adrenal, no se pueden extrapolar las estimaciones epidemiológicas porque solo se tiene, en específico, información de la población adulta.8 No obstante, un estudio que evaluó la incidencia de la crisis adrenal en niños con insuficiencia adrenal tratada encontró que se presentaban 6,5 eventos/100 pacientes-año, pero solo en pacientes con hiperplasia adrenal congénita, por lo que resulta incierta la frecuencia de presentación en otras etiologías de la insuficiencia adrenal en niños.9
La insuficiencia adrenal secundaria usualmente es menos severa que la primaria porque existe conservación de la producción de los mineralocorticoides. Los recién nacidos presentan ictericia colestásica prolongada, falla de crecimiento, hipoglucemia grave asociada a convulsiones e, incluso, coma.3 Por otra parte, los electrolitos séricos no suelen estar alterados porque no afecta la producción de aldosterona. Su debut puede ser la crisis adrenal: complicación más grave de esta entidad. En niños se caracteriza por un deterioro agudo del estado de salud debido a un trastorno hemodinámico agudo o un desequilibrio electrolítico (hiponatremia, hipercalemia, hipoglucemia sin otra causa aparente).3,10 Es común que la crisis adrenal no se sospeche y su desenlace resulte fatal si no se trata adecuadamente porque puede confundirse con otras causas de choque como sepsis, bacteriemia, gastroenteritis.3,9 Por esta razón, el objetivo de este trabajo fue examinar el caso de una paciente con insuficiencia adrenal central que debutó con una crisis adrenal congénita.
Presentación del caso
Recién nacida a término (39 semanas de gestación), parto vaginal, fruto de segunda gestación, peso al nacer de 2860 g, talla al nacer 52 cm y Apgar de 8 y 10, padres no consanguíneos y antecedente de hermano fallecido en la primera semana de vida por aparente infección bacteriana. Se hospitalizó a los nueve días de vida por presentar emesis progresiva que no respondía al tratamiento con antieméticos, y posteriormente, desarrolló episodios de apnea. Ingresó a servicios de urgencias con deshidratación severa y estuporosa, glucometría 34 mg/dL, con presión arterial 45/30 mmHg (en percentil < 5 para la edad), frecuencia cardíaca 168 lpm, frecuencia respiratoria 68 rpm, temperatura de 36,6 °C, saturación de oxígeno 85 %, peso 2,3 gr (P/T: -2,5), talla 52 cm (T/E: 0,71) y PC 35 cm (PC/E: 0,22). Al examen físico no se encontró soplo cardíaco; se reportó abdomen sin masas y genitales femeninos normoconfigurados.
Recibió reanimación hidroelectrolítica con solución salina normal (SSN) bolo de 40 cc que requirió repetir; además, se administró un bolo de dextrosa al 10 %, y ante hipoglucemia refractaria se necesitó mantener un flujo metabólico de 14 mg/kg/min. Los laboratorios informaron acidosis metabólica severa con brecha aniónica aumentada, hipoglucemia central, hiponatremia y falla renal prerrenal. Por la sospecha de sepsis neonatal tardía, se inició tratamiento con antibióticos (ceftriaxona) con previa toma de urocultivo y hemocultivos. Se trasladó para la unidad de cuidado intensivo pediátrico en la que precisó tratamiento vasopresor con dobutamina y milrinona. Sin embargo, ante el choque refractario a catecolaminas se administró hidrocortisona a 100 mg/m2/día en infusión continua con posterior mejoría. Se mantuvo la hidrocortisona durante tres días y se disminuyó a 30 mg/m2/día.
Una vez estable se tomó una muestra crítica con hipoglucemia-insulina bajo estricta supervisión, que registró una deficiencia de cortisol de origen central. Adicionalmente, se realizaron estudios para evaluar hormonas de la adenohipófisis y no se encontraron alteraciones, por lo que se confirmó el diagnóstico de deficiencia aislada de cortisol de origen central (tabla).
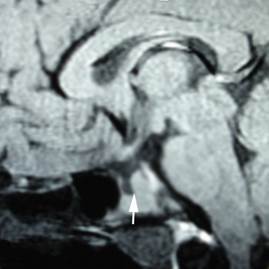
Posteriormente, se solicitó una resonancia magnética nuclear (RMN) con Gadolinio de región hipotálamo hipofisaria y se observó hipoplasia adenohipofisaria (fig.).
Se continuó la administración de hidrocortisona a 30 mg/m2/día vía oral con evolución favorable; egresó a los 16 días y continuó en seguimiento por consulta externa con controles periódicos por endocrinología pediátrica y pediatría.
Tabla Valores de los resultados de laboratorio de la paciente con los valores de referencia
| Laboratorios | Resultado | Valores de referencia |
|---|---|---|
| Hormona de crecimiento | 10,4 ng/ml | > 10 ng/mL |
| Cortisol | 2,3 mcg/dL | 5-25 mcg/dL |
| 17 alfa Hidroxiprogesterona | 2,8 ng/mL | < 5 ng/mL |
| ACTH | 10,2 pg/mL | 20-60 pg/mL |
| Insulina basal | 3,1 U/mL | 5-15 U/mL |
| Prolactina | 21,4 ng/mL | 4,5-56 ng/mL |
| T4 libre | 18,4 pmol/L | 15-34 pmol/L |
Discusión
La IA puede pasar desapercibida por su sintomatología inespecífica como pérdida de peso, fatiga, mialgia, dolor abdominal recurrente, entre otras2. Cuando se presenta la crisis adrenal aparecen síntomas como hipotensión de difícil manejo, deshidratación, náuseas intensas, confusión, hiponatremia, hipoglucemia o trastornos del potasio; de ahí su alta morbilidad y mortalidad.2,10
En la IA primaria se presenta alteración de los mineralocorticoides, a diferencia de las de origen central, en la que la renina evita la hiponatremia; no obstante, los pacientes pueden presentar hiponatremia dilucional sin trastornos del potasio debido a que el cortisol ayuda a regular la excreción de agua libre.3,11
La paciente presentó hipoglucemia temprana de difícil atención médica que es frecuente encontrarla en la IA central congénita aislada.12 Puede acompañarse de choque sin respuesta a catecolaminas, como sucedió en el caso descrito, con posterior mejoría después de la administración endovenosa de hidrocortisona a dosis de estrés. En la paciente, asumir el antecedente de la muerte neonatal temprana del hermano orientó el diagnóstico hacia la deficiencia de ACTH congénita aislada, la cual, en la mayoría descritas hasta el momento, resultan secundarias a mutaciones en el gen TPIT;5,6 desafortunadamente no fue posible realizar su determinación por no disponer de laboratorio de biología molecular.
Ante la sospecha de crisis adrenal se recomienda la toma de muestra del cortisol plasmático y de la ACTH, e iniciar el tratamiento independiente de los resultados.6 Una vez se estabilice el cuadro agudo se deben realizar pruebas dinámicas para evaluar la funcionalidad del eje hipotálamo-hipófisis-suprarrenal, en especial cuando los valores plasmáticos no permiten la confirmación diagnóstica.2,3 En la paciente, una vez estable y con supervisión estricta, se realizó el test de hipoglucemia-insulina para evaluar la función del eje; aunque este se considera el gold standard para el diagnóstico de IA secundaria, puede causar convulsiones inducidas por hipoglucemia e hipocalemia posterior al tratamiento con infusión de glucosa, por lo que se suele reservar como alternativa a otras pruebas más seguras.3,12
Una vez se diagnostica la IA secundaria resulta necesario revisar los demás ejes hipofisarios porque la deficiencia de ACTH aislada se toma como un diagnóstico de exclusión dada su escasa frecuencia. Adicionalmente, se debe realizar una RMN con Gadolinio de la región hipotálamo hipofisiaria para identificar tumores u otras lesiones y considerar que las causas más comunes resultan los procesos neoplásicos, infiltrativos, infecciosos o el tratamiento de estos (radioterapia o cirugía).1,2 En la resonancia de la paciente se observó al corte sagital T1 la glándula hipofisaria anterior con un tamaño reducido, pero la imagen hiperintensa que correspondió a la neurohipófisis se mostraba en concordancia con la ausencia de poliuria.
Existe un caso en el que un paciente con hipoplasia adenohipofisaria congénita aislada mostraba sintomatología similar, pero con complicaciones como convulsiones y dificultad para la regulación térmica; además, tenía la hipoplasia hipofisaria con otras alteraciones hormonales de la adenohipófisis que explicaban su sintomatología, para la cual requirió tratamiento con hidrocortisona y levotiroxina.13
El tratamiento de la crisis adrenal se basa en la reposición de líquidos endovenosos isotónicos, glucosa intravenosa y dosis altas de hidrocortisona. En la población pediátrica se puede iniciar con un bolo entre 25 y 50 mg/m2 de área de superficie corporal seguido de 75-100 mg/m2 por día en infusión continua y en el buretrol cada 6 h.1,5,8,10) Una vez resuelto el cuadro agudo, el tratamiento continúa con las dosis recomendadas de 10-20 mg/m2/día dividida en 2 o 3 dosis. Actualmente, la hidrocortisona resulta el glucocorticoide de elección por su perfil de seguridad, se considera la más fisiológica y la que menos compromete el crecimiento.12,14
En conclusión, la crisis adrenal debe sospecharse ante cuadros agudos con choque hemodinámico persistente, los cuales no respondan al tratamiento convencional y que pueden estar asociado a hipoglucemia de difícil control. Es fundamental el uso de hidrocortisona a dosis de estrés y tener en cuenta la hipoplasia hipofisaria como una causa de insuficiencia adrenal en neonatos. El tratamiento de la IA debe ser oportuno teniendo en cuenta factores desencadenantes para evitar la crisis adrenal que presenta una alta mortalidad.

