










 Servicios personalizados
Servicios personalizados Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de Axenfeld-Rieger lleva el nombre del oftalmólogo alemán Theodor
Axenfeld que estudió los trastornos del segmento anterior ocular y del austríaco Herwigh Rieger.1 Fue descrito en 1920 por Axenfeld como anomalía de Axenfeld y en 1934 ya Rieger lo describe acompañado de anomalías del iris y alteraciones no oculares como dentarias y faciales.2
Es una enfermedad de origen genético, autosómica dominante en el 75 % de los casos, de alta penetrancia y expresividad variable que tienen en común su origen en la cresta neural durante el desarrollo embrionario y fetal, por migración anómala, con detención del desarrollo de los tejidos derivados de las células de la cresta neural durante el tercer trimestre de gestación, lo que explica las anomalías oculares, así como otras anomalías extraoculares, especialmente las dentales y faciales.1)
Los defectos oculares clásicos del síndrome de Axenfeld-Rieger incluyen hipoplasia del iris, adherencias iridocorneales, corectopia, policoria, embriotoxon posterior, y otros rasgos menos frecuentes como cataratas, desprendimiento de retina y microcórnea. Las anomalías sistémicas asociadas incluyen las malformaciones faciales (telecantus, hipoplasia del maxilar), anomalías dentarias y piel periumbilical redundante entre otras. Aproximadamente la mitad de los individuos afectados desarrollan glaucoma.3
Se describen un caso clínico con el objetivo de mostrar la asociación del Síndrome de Axenfeld-Rieger y el glaucoma en la edad pediátrica, así como su manejo y evolución durante 10 años.
Presentación del caso
Paciente masculino de 18 años de edad con diagnóstico de síndrome de AxenfeldRieger, realizado por genética médica. A los dos años de edad fue intervenido quirúrgicamente por oclusión intestinal y se realizó reconstrucción umbilical. Como antecedente familiar la madre padece hipotiroidismo, no se reportan antecedentes familiares del síndrome, ni de otras entidades oftalmológicas o generales.
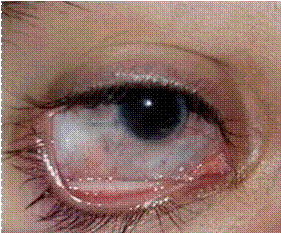
Es referido a la consulta de oftalmología pediátrica del Instituto Cubano de Oftalmología Ramón Pando Ferrer en el año 2011, a los 8 años de vida, por presentar escleras azules, microcórnea, embriotoxon posterior, aniridia parcial (Fig. 1), atrofia sectorial del iris, edema corneal y pigmentos a nivel del cristalino de forma bilateral. En el examen del fondo de ojo se halló papilas con excavación de 0,6 en ojo derecho y 0,7 en ojo izquierdo, con disminución del anillo neurorretiniano en sector inferior y temporal, y cambios del epitelio pigmentario retinal a nivel ecuatorial asociado a cifras elevadas de presión intraocular en ambos ojos, por lo que se decide iniciar tratamiento médico.

Las cifras tensionales se mantenían altas a pesar de encontrarse con triple terapia de tratamiento hipotensor tópico a dosis máxima. Por esta razón se decide realizar trabéculo-trabeculectomía en ambos ojos. Al mes de la cirugía se constata un aumento de las presiones intraoculares del ojo izquierdo, se inició tratamiento médico que fue efectivo durante un año. Tras nueva descompensación se realiza trabéculo-trabeculectomía con mitomicina C en sector temporal, manteniendo cifras de presión intraocular dentro de límites normales durante cinco años. Al mostrar nuevas cifras tensionales elevadas se decide implantar válvula de Ahmed con trabéculo-trabeculectomía con mitomicina C. Hasta la fecha se mantiene compensado con tratamiento médico con timolol al 0,5 % una gota cada doce horas.
En el 2018 se implanta dispositivo de molteno con trabéculo-trabeculectomía con mitomicina C en ojo derecho. No presentó complicaciones en postoperatorio inmediato, solo hipertensión ocular de la quinta semana, lo cual resolvió con tratamiento médico al mes del comienzo de la hipertensión ocular, manteniéndose compensado durante dos años. Posteriormente mostró cifras elevadas que no compensaban con tratamiento médico, por lo que se decide su estudio y nueva estrategia quirúrgica.
En esta ocasión la agudeza visual sin cristales fue de 0,4 en ojo derecho y de 0,5 en ojo izquierdo y la agudeza visual mejor corregida fue de 0,7 en ojo derecho y de 1,0 en ojo izquierdo, con un defecto refractivo de -11,00 -1,25 x 30° en ojo derecho y de -11,00 -2,00 x 55° en ojo izquierdo.
Al examen oftalmológico de los anexos oculares se aprecia escleras azules en ambos ojos. En el segmento anterior se observa microcórnea, embriotoxon posterior, aniridia parcial, atrofia sectorial del iris en ambos ojos. Los tubos de ambos implantes en posición, sin contacto con iris ni endotelio. Los medios refringentes no muestran edema corneal, solo la presencia de pigmentos a nivel del cristalino bilateral.
En el examen fundoscópico se encuentran papilas con excavación 0.7 en ambos ojos, con disminución del anillo neuroretiniano en sector inferior y temporal, con cambios del epitelio pigmentario retinal a nivel ecuatorial.
Los valores de paquimetría central fueron de 677μm ojo derecho y 655 μm ojo izquierdo. Los valores de presión intraocular corregidas fueron de 45 mmHg en ojo derecho y 21 mmHg ojo izquierdo.
Al examen por gonioscopía se observa una línea de Schwalbe prominente desplazada en sentido anterior, inserción anterior del iris en sector inferior y superior, procesos iridianos que llegan hasta el trabéculo pigmentado.
En los estudios de campo visual automatizado umbral muestran un aumento de la mancha ciega hacia superior en ojo derecho y un defecto arciforme superior en ojo izquierdo. Ultrasonográficamente no se encontraron alteraciones.
Dentro de las alteraciones no oftalmológicas se aprecia hipoplasia del maxilar inferior, alteraciones dentarias y alteraciones osteomioarticulares (Fig. 2).

Por los hallazgos encontrados al examen físico se decide realizar revisión con aguja (calibre 25 G) siguiendo los protocolos de tratamiento. Se penetró por una zona alejada de la zona filtrante, se diseccionó el tejido hasta llegar al plano escleral, desbridando las adherencias y escindiendo totalmente la cápsula de Tenon hasta visualizar la formación de la ampolla filtrante y salida de acuoso al exterior a través del orificio conjuntival. Se administró 1 décima de trabéculotrabeculectomía con mitomicina C al 0,01 % subconjuntival (Fig. 3).
En el postoperatorio inmediato la presión intraocular corregida fue 10 mmHg ojo derecho y de 18 mmHg en ojo izquierdo con tonómetro de aire.
Al mes los valores de presión intraocular alcanzaron 17 mmHg ojo derecho y 20 mmHg ojo izquierdo con tratamiento con Timolol al 0,5 % en ojo izquierdo. No se encontraron variaciones evolutivas en el examen de fondo de ojo. Manteniendo cifras de presión intraocular dentro de límites normales hasta la fecha.
Discusión
El síndrome de Axenfeld-Rieger es una rara enfermedad congénita del desarrollo ocular, que afecta al segmento anterior del ojo y presenta también alteraciones extraoculares acompañantes. Pertenece al grupo de las llamadas disgenesias del segmento anterior, previamente conocidas como disgenesias mesodérmicas.1 Estos cambios morfológicos conducen a su asociación con glaucoma debido a anomalías del desarrollo angular donde hay un aumento de la resistencia al flujo del humor acuoso provocando hipertensión ocular. Frecuentemente las alteraciones oculares coexisten con otros hallazgos sistémicos que caracterizan el síndrome.3)
En la anomalía de Axenfeld solo se observa embriotoxon posterior y unas bandas sinequiadas del iris periférico hacia esta línea con alteraciones del ángulo. La anomalía de Rieger presenta las alteraciones centrales del iris como hipoplasias estromales, corectopias y se suman las alteraciones de la anomalía de Axenfeld.
El síndrome de Axenfeld-Rieger tiene las alteraciones de la anomalía de Rieger más las alteraciones sistémicas.4)
El síndrome de Axenfeld-Rieger tiene una prevalencia mundial de 1:200,000.5 La transmisión es usualmente dominante para el grupo de Axenfeld-Rieger, pero puede ser esporádico. Se han identificado cinco sitios cromosómicos (4q25, 6p25, 11p13, 13q14, 16q24) y 3 genes de factor de transcripción. Dos de estos genes, PITX2 y FOXC1, que se vinculan a los cromosomas 4q25 y 6p25 respectivamente, son los más frecuentemente afectados. (3
Las alteraciones oculares observadas afectan principalmente al iris, córnea y cámara anterior. Los cambios a nivel del iris son de gran importancia al establecer el diagnóstico del síndrome de Axenfeld-Rieger, e incluyen hipoplasia, corectopia y policoria. El paciente presenta aniridia parcial bilateral y atrofia sectorial del iris.
La línea de Schwalbe es prominente y se sitúa anteriormente (embriotoxon posterior). Aparece como una línea blanca en la córnea posterior cerca del limbo y puede observarse en lámpara de hendidura o en la gonioscopía. El embriotoxon posterior se encuentra en la mayoría de los pacientes, pero aproximadamente 15 % de la población general lo presenta, sin riesgo de desarrollar glaucoma. Cuando se identifica en un paciente con disgenesia del segmento anterior, la primera consideración debe ser síndrome de Axenfeld-Rieger. La ausencia de alteraciones corneales como megalocórnea, esclerocórnea u opacidades corneales, son criterios para diferenciar el síndrome de Axenfeld-Rieger de otras disgenesias del segmento anterior.6
A nivel del ángulo se aprecia el embriotoxon posterior asociado a adhesiones iridocorneales. Por detrás de ellas, se encuentra un ángulo abierto, con el trabéculo visible y el espolón escleral parcialmente oculto por una inserción iridiana en la porción posterior de la red trabecular.7
El glaucoma es el problema ocular más serio en estos pacientes, frecuentemente bilateral, se encuentra presente en 50 % de los casos y a veces desde el nacimiento. Puede aparecer durante la niñez, pero es más común durante la adolescencia o al principio de la madurez. Se caracteriza por ser de difícil manejo y tener un pronóstico visual reservado.8
Son posibles otros trastornos oculares: catarata, degeneración macular, hipoplasia del nervio óptico, desprendimiento de retina e hipermetropía. Nuestro paciente presenta pigmentos iridianos a nivel del cristalino, además de cambios del epitelio pigmentario retinal a nivel ecuatorial.
Dentro de las manifestaciones sistémicas se describen las anomalías faciales como la hipoplasia maxilar y mandibular, aplanamiento de la base de la nariz, telecantus e hipertelorismo. Dentro de las anomalías dentarias se encuentran la microdontia, anodontia, oligodontia (predominantes en los incisivos superiores). Las anomalías óseas y articulares están caracterizadas por apófisis clinoides posteriores prominentes, centro pseudoepifisario del metatarso y metacarpo, osificación irregular de la cabeza femoral y de las epífisis distales tibial y femoral, segundo y quinto metatarsos cortos. En el sistema cardiovascular se incluyen los defectos de los tabiques interauriculares e interventriculares. En el sistema nervioso central se puede detectar hipoacusia neurosensorial, hidrocefalia, calcificaciones leptomeningeas, retraso mental leve y en algunos casos, se observa hipospadia en pacientes masculinos.9 En el caso clínico se aprecia hipoplasia del maxilar inferior, alteraciones dentarias, defectos osteomioarticulares y fue intervenido quirúrgicamente de pliegue umbilical redundante .
El glaucoma constituye una de las principales causas de mala visión en pacientes con Síndrome de Axenfeld-Rieger. La elevación de la presión intraocular es debido a la iridogoniodisgenesia y a las alteraciones de las estructuras angulares. Este tipo de glaucoma con frecuencia es refractario a tratamiento medicamentoso, por lo que requiere de un tratamiento quirúrgico para obtener el control de las presiones intraoculares.10)
La goniotomía y la trabeculotomía se consideran tratamientos con poca efectividad. La trabeculectomía es el procedimiento quirúrgico de elección y sus resultados son comparables a los alcanzados con otras formas de glaucoma, que comprometen a pacientes en un rango de edad similar. Desafortunadamente como muchos son niños, tienen un pronóstico más reservado respecto a la utilidad de esta cirugía por la respuesta fibrótica exagerada, característica de su grupo de edad.7
En la actualidad existen autores que consideran los implantes valvulares como la primera elección quirúrgica. Sobre todo en los síndromes iridocorneales como el síndrome de Axenfeld-Rieger, por su elevado índice de fracaso mediante cirugía convencional.11)
Dentro de los dispositivos de drenaje el más empleado para uso pediátrico es el implante valvular de Ahmed de adulto, los modelos pediátricos están indicados en niños menores de seis meses o en ojos con biometrías menores de 22 mm. Los niños tienen el dispositivo por un mayor tiempo por lo que se hace necesario que la tasa de éxito sea mayor. No obstante, pueden realizarse procedimientos alternativos médicos o quirúrgicos, incluso implantes adicionales, también tubos extensores cuando es necesario debido a las complicaciones propias del crecimiento del ojo. El modelo más empleado es el FP7 de adulto de plato flexible, reduce la aparición de quistes en la cápsula de tenon.12)
En general, los dispositivos de drenaje ofrecen la mejor tasa de éxito a largo plazo en glaucomas pediátricos, aunque la tasa de complicaciones es elevada, más incluso que en adultos, y se relacionan sobre todo con la hipotonía y con el propio tubo. Por eso, aunque la técnica difiere poco de la que se realiza en el adulto, se deben tomar todas las precauciones para evitar estas complicaciones.13
Alrededor de la cuarta semana el implante se rodea de una cápsula fibrosa compuesta por fibroblastos, colágeno y glicosaminoglicanos. Este proceso de fibrosis puede estar exacerbado dependiendo de la condición precedente, y esto da lugar a la encapsulación de la ampolla. Para muchos autores forma parte de la evolución normal posquirúrgica, lo que guarda relación con la llamada fase hipertensiva, presente al final del primer mes en 20 % a 40 % de los casos. Para otros, puede estar presente hasta en el 82 % de los pacientes.14)
El paciente además de estar en edad pediátrica lo cual constituye un factor de riesgo para el fallo quirúrgico por la predisposición a la fibrosis posquirúrgica presenta la condición de presentar el síndrome en cuestión, lo cual también predispone a la fibrosis y a una evolución refractaria del glaucoma asociado, por lo que se decide dada la descompensación de las presiones intraoculares realizar revisión con aguja (calibre 25 G) y administración de mitomicina C en el área quirúrgica con una evolución favorable.
Los signos que identifican el fallo de la filtración por ampolla encapsulada son el incremento de la presión intraocular en ausencia de ostium quirúrgico obstruido, y la presencia de quiste de Tenon. Este último se caracteriza por una cavidad llena de líquido, muy elevada, localizada, de consistencia firme, hipertrófica y con vasos ingurgitados en su superficie. La cavidad atrapa al humor acuoso y evita la filtración. Desde 1941, varios autores han propuesto la revisión con aguja (cistitomía) como una técnica sencilla y segura para el rescate de ampollas encapsuladas. Esta ofrece buenos resultados, incluso años después de una cirugía filtrante, y puede ser aplicable en cirugías fallidas de glaucomas congénitos. La revisión con aguja se combina al uso de fármacos en inyección subconjuntival que modulan la cicatrización. Estos son los antimetabolitos, los inhibidores de los factores de crecimiento (bevacizumab, interferón) y los espaciadores (viscoelásticos). El uso de antimetabolitos (mitomicina C y 5-fluorouracilo) es el método más común y antiguo. Otros estudios en el conejo e investigaciones in vitro han demostrado una reducción en la cicatriz posoperatoria y la falla de la ampolla con el uso de bevacizumab solo o como un complemento a 5-FU.15) Hasta la última evaluación realizada, nuestro paciente mostraba una ampolla funcional y un adecuado control tensional.
A pesar de los avances en el diagnóstico y tratamiento del glaucoma, aún no existe una terapia efectiva y única para el tratamiento de casos complejos como son los pacientes con síndrome de Axenfeld-Rieger. Se considera de gran importancia resaltar el manejo adecuado de estos pacientes y su evolución médica, por el reservado pronóstico visual que presentan.

