










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de Robinow (SR) o síndrome de facies fetal es una enfermedad genética rara que se caracteriza por un acortamiento de las extremidades y anomalías en la cabeza, la cara y los genitales externos. Fue descrito por Meinhard Robinow y sus colegas en 1969, de donde proviene su nombre original; su segundo nombre se debe a los rasgos faciales semejantes a los de un feto de 8 semanas.
Esta entidad congénita tiene una prevalencia mundial de 1 por cada 500 000 niños, con igual incidencia por sexo y heterogeneidad genética, pues puede ser heredada de la madre o el padre y no se vincula a los cromosomas de sexo.1
Se han identificado diferentes características y componentes genéticos asociados, en específico el gen ROR2 (localizado en el cromosoma 9 [9q22]), con un modelo de heredabilidad autosómico recesivo, que muestra datos semiológicos más marcados y mayores alteraciones esqueléticas, mientras que las formas dominantes son las más comunes y están determinadas por acortamiento de las extremidades (entre leve y moderado) y anomalías en la cabeza, la cara y los genitales externos; estas formas dominantes son causadas por mutaciones sin sentido en el gen WNT5A (localizado en el cromosoma 3p14.3) o en las proteínas DVL1 o DVL3.2
Según los Informes Periódicos de Orphanet,3 en su Serie de Enfermedades Raras, hasta enero del 2020 se habían descrito unos 200 casos con síndrome de Robinow, de los cuales 100 eran autosómicos dominantes y los otros 100 autosómicos recesivos, sin diferencias en cuanto al sexo. Estos fueron registrados en EE.UU., países árabes, Turquía, República Checa, Eslovaquia, el subcontinente indio y Brasil; en otras regiones es desconocida la entidad clínica.
El síndrome de Robinow afecta la estructura corporal de forma generalizada, especialmente el área craneofacial (macrocefalia, hipoplasia facial, micrognatia e hipertelorismo ocular o presencia de una separación anormal o excesiva de las órbitas oculares), la cavidad bucal (alteraciones dentarias e hiperplasia gingival), el sistema musculoesquelético (estatura baja, alteraciones vertebrales, comúnmente la escoliosis), los genitales (más evidente en los varones con la presencia de hipoplasia genital o de criptorquidia), y los riñones (hidronefrosis [con mayor frecuencia: 25 %], displasia quística del riñón, duplicación renal). También pueden aparecer, con menos periodicidad, anomalías y alteraciones cardíacas. Respecto al área neurológica, la inteligencia y las funciones cognitivas presentan un nivel normal en la mayoría de los casos, aunque puede existir retardo mental en hasta 20 % de los afectados.4,5,6
El diagnóstico prenatal se establece a partir de la semana 19 de embarazo mediante la ecografía para el estudio de la longitud de diferentes componentes óseos si existe riesgo genético, pero es difícil determinar la gravedad del síndrome.7
Por su parte, el diagnóstico posnatal es fundamentalmente clínico, pues se basa en la observación de la evolución clínica, el estudio de la historia médica individual y familiar y el examen físico exhaustivo. Algunos hallazgos deben ser confirmados a través de pruebas radiológicas, especialmente las anomalías óseas (en extremidades, cráneo, columna vertebral).
De forma general, el pronóstico de estos pacientes es bueno y la gravedad depende de la aparición de complicaciones, fundamentalmente cardíacas, pulmonares o renales, que condicionan su esperanza de vida.
En otro orden de ideas, existen pocos datos sobre la incidencia de enfermedad renal crónica (ERC) en la niñez y la adolescencia. Al respecto, en Europa se registran aproximadamente de 10 a 12 pacientes por cada millón de la población infantojuvenil y prevalencias de alrededor de 59-74 por cada millón de ese grupo poblacional. Es más frecuente en varones (63,3 %) y, en cuanto a la raza, en Norteamérica la incidencia es de 2-3 veces mayor en niños afroamericanos. Las anomalías estructurales resultan ser la causa de ERC en más de la mitad de los niños afectados (57 %), seguidas de las enfermedades renales quísticas y hereditarias (16 %), las enfermedades vasculares (9,4 %) y las glomerulopatías primarias o secundarias (5,1 %).8
Según las guías KDIGO de 2012,8 la ERC tiene los siguientes criterios durante un periodo mayor de 3 meses: filtrado glomerular disminuido (FG menor de 60 ml/min/1,73 m2) y/o presencia de uno o varios marcadores de daño renal (albuminuria aumentada, anomalías del sedimento urinario -electrolíticas, por trastornos tubulares o detectadas histológica o imagenológicamente- y antecedente de trasplante renal).
Caso clínico
Se describe el caso de un paciente de 18 años, quien fue asistido en el Servicio de Nefrología del Hospital Infantil Docente Norte Dr. Juan de la Cruz Martínez Maceira de Santiago de Cuba por presentar síndrome hidropígeno. Las pruebas revelaron una disminución marcada de la función renal, por lo cual requirió terapias sustitutivas y se consideró como un fallo renal agudo de causa multifactorial. Al persistir por más de tres meses esta condición, se diagnosticó como insuficiencia renal crónica en fase terminal dependiente de hemodiálisis.
Como antecedentes patológicos personales, el paciente refirió que había sido operado de hipertelorismo en su primer año de vida, además padecía pangastritis urémica y úlcera gastroduodenal. Señaló que no tenía ningún antecedente patológico familiar ni perinatal, tampoco traumatismos ni hábitos tóxicos.
En busca de la causa de la enfermedad, se le realizaron varios exámenes.
Examen físico
Facies: Ojos separados (hipertelorismo), frente ancha y plana, rasgos faciales toscos. Perfil facial plano (fig. 1).
Cuerpo: Escoliosis con desviación a la derecha.
Faneras: Hipoplasia ungueal.
Sistema arterial: Fístula arteriovenosa en el brazo izquierdo, que funcionaba bien, sin complicaciones.

Exámenes imagenológicos
Tomografía axial computarizada de cráneo y órbitas: Signos de atrofia cerebral.
Fondo de ojo: Palidez del nervio óptico; el resto normal.
Rayos X de columna lumbar: Escoliosis lumbar de concavidad derecha y espina bífida S1.
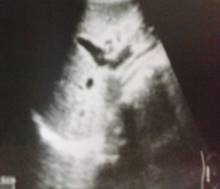
Ecografía abdominal: Hepatoesplenomegalia, riñones con características propias de ERC (fig. 2).
Ecografía Doppler hepática: Vena porta de 16 mm, con flujo bidireccional y gráfica espectral sugestiva de hipertensión portal (fig. 3).

Se interconsultó con otras especialidades, entre ellas la de Genética Clínica, por los trastornos dismórficos encontrados en el examen físico. Se concluyó que se trataba del síndrome de Robinow, o síndrome de facies fetal, concomitante con insuficiencia renal crónica de causa no precisada, hipertensión portal y glaucoma.
Tratamiento aplicado
Primeramente, se indicó tratamiento depurador de hemodiálisis 3 veces en la semana por medio de la fístula arteriovenosa.
Luego de efectuados los exámenes complementarios de laboratorio, se añadieron suplementos dietéticos para paliar las alteraciones del metabolismo fosfocálcico y la anemia, con agentes estimulantes de la eritropoyesis (eritropoyetina humana recombinante EPO rHu) y suplementos de hierro; además, se decidió que el paciente recibiera apoyo psicológico.
El paciente permaneció en hemodiálisis por un periodo de 2 años hasta que se logró estabilizar su condición de salud para poder efectuar un trasplante de riñón (que recibió de un donante fallecido). Después de la operación, la función renal se mantuvo normal durante 6 meses, pero luego fue necesario aplicar nuevamente hemodiálisis; tratamiento que recibe en la actualidad.
Comentarios
El síndrome de Robinow es una entidad clínicamente polimórfica y de rara aparición.9 En la bibliografía médica foránea10 se registra una mayor frecuencia de las anomalías anatómicas y funcionales, sobre todo bucales y óseas; sin embargo, apenas se han publicado estudios genéticos que demuestren la mutación causante de alteraciones estructurales renales, que son las menos usuales, solo se halló una serie de casos donde los pacientes que recibieron trasplante de riñón presentaban insuficiencia intelectual por diferentes causas, una de ellas era el síndrome de Robinow, en tanto, la insuficiencia renal era de origen desconocido. El actual caso clínico tuvo características similares a las anteriores, pues al paciente se le diagnosticó la insuficiencia renal ya en estadios avanzados sin encontrar una causa evidente y, además, se requirió tratamiento depurador con hemodiálisis durante un largo periodo hasta que se logró estabilizar su estado físico para realizar un trasplante de riñón.
Actualmente no existe una cura para las personas con síndrome de Robinow. En los informes la terapia se centra en la resolución de las complicaciones clínicas, mientras que las alteraciones bucodentales son corregidas con el tratamiento adecuado para ello, las musculoesqueléticas suelen mejorarse con terapia física, colocación de prótesis o corrección mediante procedimientos quirúrgicos, y las alteraciones cardíacas y genitales son disminuidas con tratamientos farmacológicos y/o quirúrgicos. También existen otros tipos de terapias novedosas que se basan en la administración de hormonas del crecimiento, para estimular el incremento de la estatura.
El diagnóstico y tratamiento precoces son fundamentales para la corrección de alteraciones estructurales y el control de las complicaciones, tanto cardíacas como renales. Así pues, se requieren equipos multidisciplinarios que, a través de la intervención terapéutica, física, social y psicológica, puedan promover el desarrollo de capacidades y habilidades en los niños afectados por este síndrome, a fin de lograr una mejor calidad de vida en ellos, sobre todo en los que poseen rasgos faciales dismórficos que dañan sus relaciones interpersonales y sociales, o en aquellos que han sido diagnosticados con enfermedad renal crónica en fase terminal y requieran tratamientos sustitutivos de la función renal (hemodiálisis y trasplante de riñón), los cuales son muy invasivos, reiterados y prolongados.

