










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Estomatología
versión impresa ISSN 0034-7507
Rev Cubana Estomatol vol.50 no.1 Ciudad de La Habana ene.-mar. 2013
REVISIÓN BIBLIOGRÁFICA
Propuesta de teorías integradoras para la cefalogénesis y sus malformaciones
Proposal of integrative theories for cephalogenesis and their malformations
Dadonim Vila Morales
Facultad de Ciencias Médicas "Finlay-Albarrán". Universidad de Ciencias Médicas de La Habana, Cuba.
RESUMEN
En las teorías embriogénicas de las malformaciones del macizo craneofacial es frecuente encontrar explicaciones simplistas, que no aluden a la biología celular, ni molecular de sus fisiopatologías. El propósito de esta investigación fue describir las teorías de la cefalogénesis y sus malformaciones craneofaciales, como requisito para proponerteorías integradoras que favorezcan su mejor comprensión. Se realizó una revisión biobliográfica sobre el tema de la cefalogénesis y las concepciones científicas actuales concernientes a las teorías de las malformaciones congénitas craneofaciales. Se realizó una revisión en Internet, en el periodo comprendido entre diciembre de 2011 y enero de 2012. Se analizaron los artículos de revistas de la Web of Sciences que trataron este tema desde el año 2006 hasta el 2012 en los buscadores Google academico, Hinari, Scopus, Scielo y Medline. Como resultado se obtuvieron 131 artículos que fueron tamizados; pero el estudio se ciñó solo a 43 artículos que enfocaron estas temáticas de manera más integral. Se consideraron reduccionitas o incompletas las teorías hasta ahora expuestas. Por tal motivo se propusieron teorías holístico-sinérgicas de la cefalogénesis y sus malformaciones.
Palabras clave: cefalogénesis, malformaciones craneales, cirugía craneofacial, anomalías maxilofaciales, teorías cefalogénicas.
ABSTRACT
Frequently is possible to find simplistic explanations regarding the embriogenics theories of craniofacial malformations, without any explanation about cellular or molecular biology of this physiopathology process. The purpose of this investigation was to provide elements to allow the understanding of cephalogenesis as to value theories of cephalogenesis and craniofacial malformations and to suggest an integrative theory. A bibliographic study was carried out about cephalogenesis, and current scientific conceptions on theories of craniofacial congenital malformations. An Internet exploring was made during December of 2011 to January of 2012. Was evaluated the Web of Sciences reports related those topics published since 2006 to 2012 in searching as: Academic Google, Hinari, Scopus, Scielo and Medline. Was obtained 131 reports as result they wich were sifted and bounded to 43 papers focused this topic in an integral way. As conclusion was brought up to date cephalogenesis, and their alterations since the knowledge of differents biological levels that participate in cranial grown. It was considered simplistic or incomplete the theories used until now and intended holistic-sinergic theories of cephalogenesis and their malformations.
Key words: cephalogenesis, cranial malformations, surgery craniofacial, anomalies maxillofacial, cephalogenics theories.
INTRODUCCIÓN
La manera en que el embrión crece y se desarrolla desde la fecundación hasta su nacimiento, ha sido desde hace muchos siglosha sido para el hombre motivo de profunda preocupación. Precisamente la escolástica prohibió su estudio creyéndolo un misterio divino, vedado al conocimiento humano. Esta concepción reticente al estudio embrionario humano generó gran retraso en la comprensión de sus esencias. Consecuentemente, desde la Edad Media y hasta finales del siglo XIX la embriología se mantuvo como una ciencia inexplorada.
Hoy día este estudio continúa motivando controversias debido a principios bioéticos que consideran al embrión como aun ser humano no nacido que merece protección y respeto. Este derecho fue consagrado por la Roma Imperial que protegía al humano no nacido y le brindaba derechos incluso de herencia.1 Es evidente que no es ético estudiar al embrión humano a expensas de su sacrificio o riesgo biológico. Por este motivo, las ciencias básicas aplicadas al estudiar los vertebrados superiores nos brindan una serie de elementos teóricos que desde la demostración en la genética y la embriología comparadas han contribuido a la comprensión sindromológica y fisiopatológica de las malformaciones craneofaciales en humanos. A su vez, el estudio del genoma humano y su mapeo, así como el desarrollo de la epigenética, han permitido la comprensión bioquímica de muchas afectaciones morfológicas craneofaciales de origen congénito.2
La estomatología ha estado un tanto alejada de la neurociencia y resulta poco frecuente el desarrollo de esta línea de investigación en los centros estomatológicos de investigación científica. En la literatura aparecen descritas muchas malformaciones de causa "idiopática", lo cual ha generado pereza en cuanto a la búsqueda de elementos teóricos que propicien una mejor comprensión de estos fenómenos embriogénicos craneofaciales.3 En las teorías embriogénicas de las malformaciones del macizo craneofacial es frecuente encontrar explicaciones simplistas, que describen parcialmente la cefalogénesis a escala tisular, y no aluden a la biología celular o molecular de sus fisiopatologías.4,5
Esa circunstancia motivó esta investigación, cuyo propósito fue describir las teorías de la cefalogénesis y de las malformaciones craneofaciales, a fin de proponer una teoría integradora que favorezca su mejor comprensión.
MÉTODOS
Se realizó un estudio de revisión bibliográfica sobre el tema de la cefalogénesis y de las concepciones científicas actuales, concernientes a las teorías de las malformaciones congénitas craneofaciales. Esta revisión se realizó en Internet, en el periodo comprendido entre diciembre de 2011 y enero de 2012. Se evaluaronrevistas de impacto de Web of Scienciesrelacionadas con estetema: Journal of CraniofacialSurgery, TheOfficialJournal of Federation of American Societiesfor Experimental Biology, Plus GeneticsJournal, Trends in Genetics, Current Top Development in Biology, TheJournal of Neurosciences, TheAnnals of New York AcademySciences, y la Revista Española de Cirugía Oral y Maxilofacial. Se analizó además, la Revista Cubana de Estomatología. Por la escasa producción científica que existe sobre esta temática la búsqueda se realizó en artículos publicados durante el periodo comprendido entre 2006 y 2012. Se accedió a estas fuentes mediante los buscadores de información y plataformas de publicación Google académico, Hinari, Scopus, Scielo y Medline. Se incluyó, además, la literatura clásica imprescindible de la especialidad, que aparece en libros o artículos de revistas publicados en fecha anterior a la referida. En la búsqueda se emplearon las palabras clave: cephalogenesis, craneal malformations, craneofacialsurgery, craneofacialanomaliestheory, maxilofacial anomalies y craneofacialsyndromes.
Como resultado de la búsqueda se obtuvieron 131 artículos, que fueron tamizados con el propósito de conservar solo los que describieran el proceso de cefalogénesis o sus teorías, así como los mecanismos de aparición de las malformaciones craneofaciales. De esta manera el estudio se circunscribió a 43 artículos que enfocaron estas temáticas de manera más integral, pues la gran mayoría no se relacionaron directamente con la temática objeto de estudio o versaron sobre el estudio pormenorizado de una sustancia morfogénica específica en casos muy particulares, sin describir los vínculos o su actividad precisa en la cefalogénesis de manera general.
Para el procesamiento de la información se elaboró un cuaderno de recolección de datos, a través de Microsoft Office Excel 2003, donde se confeccionó un documento que recogió todas las revistas analizadas y la cantidad de artículos de esta temática encontrados en ellas. La información se procesó a través del paquete estadístico SPSS. Al evaluar el comportamiento de los artículos respecto a su representatividad en las diferentes revistas científicas donde fueron publicados, 51,2 % de los artículos consultados correspondió a la Journal of CraneofacialSurgery, lo que resultó estadísticamente significativo al aplicarle el estadígrafo chi cuadrado (p= 0,0003). Los demás artículos estuvieron distribuidos de manera dispersa y no significativa entre las otras revistas. Esto demuestra el protagonismo de la mencionada revista en lo referente a la temática objeto de estudio. Las restantes revistas de cirugía maxilofacial consultadas en la Web of Sciencies no contenían artículos directamente relacionados con la cefalogénesis o con teorías relativas a ella, aunque muchos sí la mencionaban.
FISIOPATOLOGÍA DE LA CEFALOGÉNESIS
FACTORES QUE INTERVIENEN EN LA FISIOPATOLOGÍA DE LAS MALFORMACIONES CONGÉNITAS
En la fisiopatología de las malformaciones craneofaciales intervienen tres elementos básicos: los factores genéticos, los factores medioambientales y los factores citoplasmáticos. Dentro de los factores genéticos se distinguen las alteraciones a nivel de los genes y del genotipo. Básicamente, las modificaciones en los genes pueden estar dadas por activación o inactivación incorrecta del ADN, lo que genera: mutaciones, heterogenicidad, pleitropía, así como la penetrancia y expresividad variables; mientras que las modificaciones en el genotipo generan aneuploidías, inversiones, translocaciones y deleciones. A nivel citoplasmático también pueden ocurrir variaciones en la biología celular, que generan malformaciones congénitas craneofaciales, relacionados mayormente a los procesos de transcripción (defecto de transcripción) o de la síntesis proteica, ya sea por alteraciones ribosomales o de incorrecta rectificación de la proteína obtenida.6,7
Por su parte, dentro de los factores medioambientales encontramos a los elementos moduladores del metabolismo celular, ya sean de índole hormonal o teratogénico externo. En la actualidad se ha avanzado en el conocimiento de la fisiopatología de las malformaciones craneofaciales ante enfermedades endocrinometabólicas de la madre, también se ha confirmado su participación incluso en malformaciones craneofaciales adquiridas; tal es el caso del hipertiroidismo juvenil, que en niños que nacen sin alteraciones craneofaciales, produce craneosinostosistirotóxica, reportada por vez primera en el 1969, por Robinson y otros, lo cual se manifiesta con un cuadro clínico de sinostosis craneales y faciales múltiples, hipertelorismo, proptosis ocular e hipoplasia cigomática que resulta ostensible a partir de los 7 años de edad.8-10
Mucho se ha hablado de los efectos teratogénicoscraneofaciales de innumerables sustancias químicas que incluyen el alcohol, las drogas, el tabaco, fertilizantes, asbesto cemento, entre otros. En estos casos resulta directamente proporcional la gravedad de la malformación con la magnitud de la exposición y la mayor inmadurez embrionaria o fetal. Varios son los elementos que intervienen como factores citoplasmáticos, pero el más relevante de ellos es el genoma mitocondrial -que no depende del ADN nuclear, pues las mitocondrias presentan su propio ADN- cuyas mutaciones pueden ocasionar malformaciones, no necesariamente relacionadas con el ADN nuclear. Las anomalías derivadas del ADN mitocondrial pueden seguir una vía de transmisión materna, paterna o no hereditaria.10-12
NIVEL CELULAR EN LA CEFALOGÉNESIS
Antes de continuar exponiendo la fisiopatología de la biología celular de las anomalías congénitas craneofaciales es importante introducir un concepto pedagógico, para la mayor comprensión didáctica de esta materia. Se trata de la instrumentación del contenido desde una de sus variantes: la "célula genética del conocimiento".13-15 Decimos que estamos ante la célula genética de un contenido determinado, cuando hemos develado un conocimiento que constituye el núcleo esencial para la comprensión de varios procesos o fenómenos particulares que dan respuesta a los problemas o interrogantes de la profesión y que se estructuran como contenidos de enseñanza al tener respuesta desde la explicación de esa estructura conceptual. Tal es el caso del concepto de "citodiferenciación", que según criterio de este autor constituye la célula genética del conocimiento en la embriogénesis y consecuentemente en la explicación de la cefalogénesis a nivel de la biología celular. Este resulta el concepto más importante, que deviene en clave para la comprensión fisiopatológica cefalogénica.
Entendemos por citodiferenciación a la capacidad que tiene la célula embrionaria y fetal de diferenciarse para dar lugar a otro tipo de célula, constituyente de un nuevo tejido, especializado para funciones específicas, lo que ocurre bajo la influencia de una programación genéticamente codificada. Este proceso puede darse ya sea a través de la estimulación celular (citoinducción) generada por una célula sobre sí misma (auto-citoinducción) o por la mediación de sustancias producidas por células adyacentes o distantes (citoinducción a distancia). Claro está que esta citodiferenciación tiene su fundamento bioquímico en el nivel molecular, pues como se verá más adelante son las moléculas específicas y genéticamente codificadas para este proceso, los elementos activadores o moduladores de la citodiferenciación.
Precisamente la citodiferenciación genera el desarrollo de nuevos tipos celulares del ectodermo, el mesodermo y el endodermo y de sus tejidos derivados.6,7,16 Un ejemplo de este fenómeno es la formación del sistema vascular del embrión, que hacia la mitad de la tercera semana y ante el aumento de las necesidades nutricionales por el embrión, dado que la difusión de nutrientes no puede abastecer las crecientes demandas energéticas de los nuevos tejidos, ocurre la proliferación de acúmulos angiogénicos aislados. Este fenómeno se genera a expensa de la cirodiferenciación de las células mesenquimáticas de la hoja esplácnica del mesodermo (presomitas en estado avanzado). Luego aparece el tubo cardíaco y de la formación de cordones celulares de angioblastos se conformará la red de vasos sanguíneos, que a posteriori por citodiferenciaciónse canalizan, dando lugar a células endoteliales que generan un epitelio plano; otras células diferenciadas de los angioblastos dan lugar a la capa media vascular y la capa adventicia aparece secundaria a la citodiferenciación del tejido conectivocircundante.7 Durante el crecimiento y desarrollo embrionario craneofacial, los vasos se modifican grandemente: de los vasos sanguíneos pequeños se originan troncos más gruesos, ocurre la desviación del riego sanguíneo de un tronco vascular hacia otro más próximo, lo que genera un cambio de dirección del flujo, de manera temporal o permanente, en dependencia de las necesidades de nutrición sanguínea en las nuevas estructuras en formación.11 Existe además citodiferenciación que genera el colapso programado de un gran vaso sanguíneo, para dar lugar a muchos vasos de menor calibre y hasta la obliteración total de algún vaso sanguíneo y su posterior apoptosis (muerte celular programada) debido a la pérdida de su importancia nutricia.17
Cuando ocurren disrupciones genéticas o alteraciones en los procesos moleculares de la citodiferenciacióncefalogénica se generará inmadurez celular de los tejidos neoformados, con incapacidad de cumplir sus funciones específicas o incluso su necrosis (proceso que difiere de la apoptosis en su mecanismo celular de ejecución y programación genética). Pueden aparecer además alteraciones en la adhesión celular, así como repulsión celular, migración y proliferación no programadas. Estos fenómenos comprometen la morfodiferenciación de órganos y sistemas de órganos, así como del fenotipo craneofacial, secundarias a la incorrecta citodiferenciación, lo que dará lugar a anomalías de número, posición, dismorfias, denominadas genéricamente como deformidades craneales, faciales o craneofaciales. 10,18
NIVEL MOLECULAR EN LA CEFALOGÉNESIS
Si bien la citodiferenciación determina la morfología tisular y su fenotipo, este proceso tiene su base en la biología molecular. Es el nivel molecular el que determina la estructuración morfológica y funcional de todos los tejidos vivos. De esto se deriva que si la citodiferenciación es "la célula genética del conocimiento" para la comprensión de la cefalogénesis, la biología molecular es "la invariante pedagógica"15,16 de todos los contenidos de las ciencias médicas, pues sin importar cuál sea la situación o el problema de la profesión médica o estomatológica que se escrute, la comprensión de la biología molecular dará respuesta a los procesos fisiológicos o fisiopatológicos objetos de estudio.
Precisamente los procesos vitales de la célula (respiración, síntesis proteica, multiplicación, etc.) se ven condicionados por múltiples procesos moleculares. La citodiferenciación también está determinada por inductores moleculares, como mencionamos anteriormente, y que son los que definen su aparición. Dentro de los elementos moleculares citoinductores se encuentran los factores de crecimiento y los receptores morfogénicos. Durante la cefalogénesis, la ausencia o retardo de las reacciones moleculares programadas y el fallo en los receptores de interacciones moleculares van a provocar alteraciones en la citodiferenciación e histodiferenciación, lo que traerá como resultado la generación de displasias, hiperplasias o hipoplasias de los tejidos en formación.10,12
A continuación expondremos los principios generales de funcionamiento de estos inductores moleculares, que a su vez son consecuencia de fenómenos epigenéticos y que presentaremos posteriormente; pues aunque la epigenética pertenece al nivel molecular de organización celular, por sus peculiaridades será tratada de manera independiente. Centraremos nuestra atención en el nivel molecular que involucra a las cadenas polipeptídicas, secundarias a la síntesis proteica celular de inductores cefalogénicos. Los máximos exponentes de estos inductores son pequeños péptidos denominados factores de crecimiento celular.16
Son muchas las familias de factores de crecimiento celulares que han sido descritas, algunas de las cuales relacionaremos a continuación. Tal es el caso de la familia del factor de crecimiento epidérmico (EGF), dentro de la cual encontramos también al factor de crecimiento transformador alfa (TGF-A), estos, como su nombre lo indican, intervienen en la citoinducción o citodiferenciación de los tejidos ectodérmicos, para la multiplicación celular epidérmica. Otras son las familia del factor de crecimiento derivado de plaquetas (PDGF), del factor de crecimiento del tejido conectivo (CTGF) y la del factor de crecimiento de los fibroblastos (FGF), que incluye al factor de crecimiento de los fibroblastos básicos (FGFB) y ácidos (FGFA); estos últimos muy relacionadas a la proliferación fibroblásticamesenquimal, quienes a su vez son pluripotenciales en su posible citodiferenciación.6,19
Una importante estirpe de estos pequeños péptidos citoinductores es la del factor de crecimiento transformador beta (TGF-B), que incluye también a la familia de las proteínas morfogenéticas óseas (BMPs) y sus activinas. Este grupo de factores tiene un importante papel en la citodiferenciación para producción y remodelado óseo durante la cefalogénesis del neurocráneo y el viscerocráneo. Estas estructuras mesenquimales óseas no podrían proliferar sin la presencia acompañante angiogénica, que permite su nutrición y multiplicación celular, así como el aporte de minerales que formarán su andamiaje arquitectural de sostén; por ello juegan un importante papel en la cefalogénesis los factores de crecimiento del endotelio vascular (VEGF).7,20,21
En los huesos medulares y en otras estructuras óseas y hematopoyéticas están muy activos los factores estimulantes de las colonias mieloides (CSF), dentro de los que se encuentran los factores estimulantes de las colonias de granulocitos-macrófagos (GM-CSF), los factores estimulantes de las colonias de granulocitos (G-CSF), losfactores estimulantes de las colonias de macrófagos (M-CSF), así como los factores de crecimiento de la eritropoyetina y la angipoyetina (ANG). No menos importancia tienen las citocinas, como las interleuquinas, entre las que se destaca la interleucina-2 (IL-2), así como los interferones alfa (IFA) y beta (IFB) y el factor de necrosis tumoral (TNF), que aunque fue descrito primeramente en la biología oncológica, se demostró a posteriori su activa participación embrionaria. Todos ellos intervienen en múltiples procesos citoinductores, citosupresores, citomoduladores, citoseñalizadores, que determinan la citodiferenciación cefalogénica.7,22
La morfología tisular también está modulada por los factores de crecimiento Tipo-insulina I y II (IGF) y por estimuladores neurotróficoscomo son el factor de crecimiento nervioso (NGF) y el factor derivado de la cresta neural (BDNF). Existen algunos factores de crecimiento menos protagónicos en la cefalogénesis como es el factor de crecimiento de los hepatocitos (HGF) y otros muy protagónicos como el factor transcripcional (TF), quien determina en gran medida el éxito de toda la producción de citoinductores, para hacer posible la citodiferenciación y la histodiferenciación embriogénicas que determinan morfodiferenciación y el fenotipo craneofacial.6,7,21
Podemos resumir que básicamente los factores de crecimiento tienen entre sus funciones celulares: la diferenciación, proliferación, apoptosis, migración y estimulación de muchos tipos celulares. Algunos factores de crecimiento pueden inducir su propia síntesis en los llamados centros de organización de factores de crecimiento, en los que tiene actividad autoinductora. Los factores de crecimiento, como son péptidos solubles en la sangre y los fluidos tisulares, actúan a través de receptores específicos de membrana, que no son más que complejos moleculares a los que se van a acoplar los factores de crecimiento. Los receptores específicos de membrana tienen un importante papel y su alteración podría interrumpir el proceso citoinductor.3,4,21 Estos receptores receptores específicos de membrana reciben señales tanto de la propia célula, denominadas señales de origen autocrino o de células adyacentes o señales de origen paracrino, o sea distancia. De estos mecanismos citoinductores pueden generarse dos fenómenos opuestos en extremo, la proliferación y la apoptosis celular. Esta última desencadena la muerte celular, con microinflamación y fagocitosis, sin que ocurra necrosis tisular; lo que a su vez la diferencia de la muerte celular genéticamente no programada.3,6,7
BREVE DESCRIPCIÓN DE LA EPIGENÉTICA DE LA CEFALOGÉNESIS
Como ya se expresó antes, aunque la epigenética es parte del nivel molecular, por ser este el momento inicial de todos los procesos embriogénicos y consecuentemente, de los cefalogénicos, hemos preferido desarrollarla en acápite aparte.
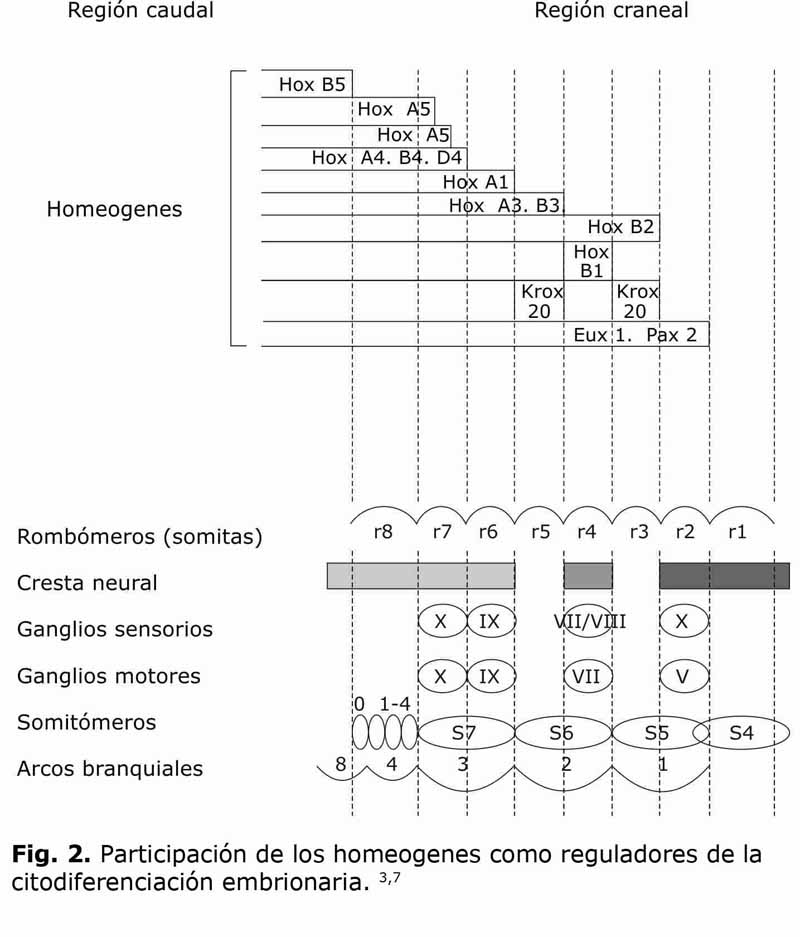
La responsabilidad del desarrollo embriogénico craneofacial ha sido atribuida mayormente a un grupo de genes denominados homeogenes. Dentro de ellos se encuentran grupos o clústeres de genes, como es el caso de los genes homeóticosHox, que junto a otros grupos de genes: Eux, Krox y Pax, controlan la especificidad regional del desarrollo y la diferenciación tisular embrionaria. Estos clústeres constituyen los sistemas proteicos reguladores de la citoinducción (SRPC) y consecuentemente de la citodiferenciación (Fig.1).7,10
El sistema regulatorio proteico Hox, tiene un segmento común cromosómico o Box denominado Homeobox, el cual está directamente involucrado en la síntesis de moléculas Hox específicas, las cuales a su vez son promotoras de un segmento de DNA específico. Existen 4 clústeres de genes Hox, (A, B, C y D,) codificados por una conformación molecular de unos 10 genes por clústeres, a su vez constituidos por 180 pares de aminoácidos base de DNA; estos a su vez codifican factores de transcripción que contienen un homeocampo (homeodomain) de 60 amino- ácidos. Cada gen Hox tiene una responsabilidad espacio-temporal específica en la inducción de expresión cefalogénica. En la región del viscerocráneo participan además otros clústeres como los Krox, que han sido tipificados hasta 20 grupos de ellos de ellos, así como los homeogenesPax, siendo el clúster más activo en este proceso el Pax 2.3,6,23
Existen diferentes clústeres de homeogenes que determinan la sincronía cronológica y morfológica de la cefalogénesis, codificando homeocampos específicos que determinan regiones cefalogénicas bien definidas (Fig. 2). Por ejemplo, la zona del I arco branquial y sus estructuras derivadas por citodiferenciación, como los procesos maxilares y mandibulares, entre otras, están determinadas por los homeogenesPax 2, Krox 20 y Hox B2. La alteración de estos sistemas proteicos reguladores de citoinducción, podrán generar malformaciones como las agrupadas en lo que antes se llamó el síndrome del primer arco branquial; que incluyen al síndrome de Treacher-Collins, el síndrome de Pierre Robin, la disostosis mandibular, el labio y paladar fisurados y el hipertelorismo, entre otras malformaciones. Otras deficiencias esqueletales derivadas de la errónea citodiferenciación del primer arco branquial son las hipoplasias mandibulares de diferentes grados, malares y las soluciones de continuidad de sus tejidos de recubrimiento, tales como las macrostomias, las microtias o acrotias, la oblicuidad antimongoloide d elas fisuras palpebrales, entre otras que luego presentaremos en una clasificación que permite su comprensión morfo-fisiopatológica. 6,7,24
Por su parte el II arco branquial es citoinducido por la actividad de los homeogenesHox B1, B2 y B3, el Krox 20 y el Eux 1, mientras que en el III arco branquial y sus estructuras derivadas intervienen los SPRC determinados por los Hox A1, A3, A5, A6 y D4 (Fig. 2). De esta manera se entiende en la actualidad los procesos complejos de la citodiferenciacióncefalogénica. Una vez comprendido su nivel celular, con la influencia de factores medioambientales, citoplasmáticos y genéticos, en relación directa con la biología celular y los procesos embriogénicos de citoinducción lo cual puede generar citodiferenciación, y con ella la migración celular, la apoptosis o el crecimiento tisular acelerado; es menester articular el nivel molecular donde los factores de crecimiento, sus receptores de membranas y los homeogenes modulan la actividad citodiferenciadora y esta a su vez tiene como componente estructural la síntesis proteica.3,7,25,26
Toda síntesis proteica se inicia en el núcleo celular, cuando el ácido desoxirribonucleico (ADN) helicoidal se estimula, en un sector específico, para desenrollarse y alinearse, y que tiene como elemento iniciador de este fenómeno a un segmento del ADN denominado segmento "cap", el que a su vez activa al segmento "líder" que le sucede y que determinará cuáles son las estructuras aminoacídicas que deberán presentarse hacia la superficie externa de la estructura molecular, las que se denominan exones y cuáles serán las secuencias que permanecerán en el interior de la misma como intrones. Este fenómeno de preparación del ADN para codificar una nueva secuencia aminoacídica en lo que será en ácido ribonucleico (ARN) de transcripción nuclear, responde precisamente a un proceso de citoinducción, ya sea autocrina (producida por la propia célula) o paracrina (producida por otras células, adyacentes o a distancia). En el caso de la autocitoinducción puede estar generada por la propia cronología nuclear predeterminada genéticamente para la producción de una sustancia específica, mientras que la citoinducciónparacrina necesita de receptores específicos de la membrana celular, que se activen para acoplarse al factor de crecimiento o péptido citoinductor.7,27,28
Una vez alineados externamente los exones, con sus secuencias aminoacídicas, ocurre la transcripción del ARN de transcripción nuclear, donde se excluyen los segmentos «cap» y del resto de la secuencia aminoacídica, quedando alineados solamente los intrones y exones. De esta estructura molecular se sintetiza a nivel de la pared nuclear, el ARN mensajero, donde aparece la base secuencial específica para la futura síntesis de la proteína inducida, sin la presencia de los intrones. En el proceso de embriogénesis va a ocurrir una traslación primaria que derivará en el posterior procesamiento y almacenamiento del nuevo producto genético, si se trata de una citodiferenciación (Fig.3). Consecuentemente, la síntesis protéica que ocurre en los ribosomas, puede ser usada como elemento estructural celular, variar su morfología y función, ser una cadena polipeptídicaautoinductora o que salga al medio extracelular con función citoinductora regional o a distancia.3,7,28
Recientemente un equipo de científicos identificó cinco genes asociados a la forma del rostro. A partir de un estudio con diez mil personas, los expertos identificaron homeogenesy algunos subtipos de ellos: PAX3,PRDM16, TP63, C5orf50 y COL17A1 implicados con las distintas formas del rostro como el color de los ojos y las características de las facciones. Según expertos a cargo de dicha investigación perteneciente al Centro Médico Universitario Erasmus, en Rótterdam, existe la probabilidad de estimar ciertas formas faciales, aunque aún está lejos la posibilidad de trazar un retrato completo. Para este estudio, los científicos emplearon imágenes de resonancia magnética de la cabeza de los voluntarios y fotografías y luego se desarrolló un estudio de asociación genética para encontrar pequeñas variaciones que se producen en personas con características faciales específicas.29
Si bien se ha esbozado la participación de los factores generales contribuyentes a las malformaciones craneofaciales, así como los procesos celulares, moleculares y epigenéticos, que las condicionan; es menester señalar, muy brevemente, los mecanismos cromosómicos más frecuentes generadores de alteraciones genéticas, que a su vez conducen a alteraciones en la transcripción del ARN y su posterior codificación en la síntesis proteica y que a su vez alteran los procesos embriofisiológicos de citodiferenciación, maduración, proliferación o muerte celular, conducentes anomalías cefalogénicas.7,10,27
ALTERACIONES EPIGENÉTICAS QUE CONDICIONAN LAS MALFORMACIONES CRANEOFACIALES
Tradicionalmente las enfermedades de causa genética han sido clasificadas categóricamente en cromosómicas (causadas por aberraciones estructurales o numéricas), monogénicas o mendelianas, multifactoriales y de origen mitocondrial. Sin embargo, esta concepción genética ha cambiado en la última década, con el empleo de la epigenéticay dicha clasificación se ha modificado, pues se han descubierto malformaciones que surgieron espontáneamente en individuos sin ningún antecedente hereditario, asociadas a mecanismos complejos que difieren de los patrones de herencia clásicos. Ejemplo de esto son las microdeleciones o microduplicacionessubmicroscópicas, las enfermedades por repetición de trinucleótidos y las malformaciones congénitas asociadas a cambios en el número de copias de secuencia codificante.10,29
Las malformaciones congénitas asociadas a cambios en el número de copias de la secuencia codificante también se denominan "desórdenes genómicos". Este concepto de desórdenes genómicos fue introducido por James Lupski en el año 1998 y se utiliza para definir aquellas condiciones que surgen por inestabilidad en la molécula de ADN y que ocasionan "rearreglos" cromosómicos que involucran regiones de uno o varios pares de megabases. En otras palabras, no son más que las malformaciones o síndromes que surgen como consecuencia de rearreglos en la molécula de ADN y que aparecen de novo en un grupo familiar o presentan un patrón clásico de herencia mendeliano.12
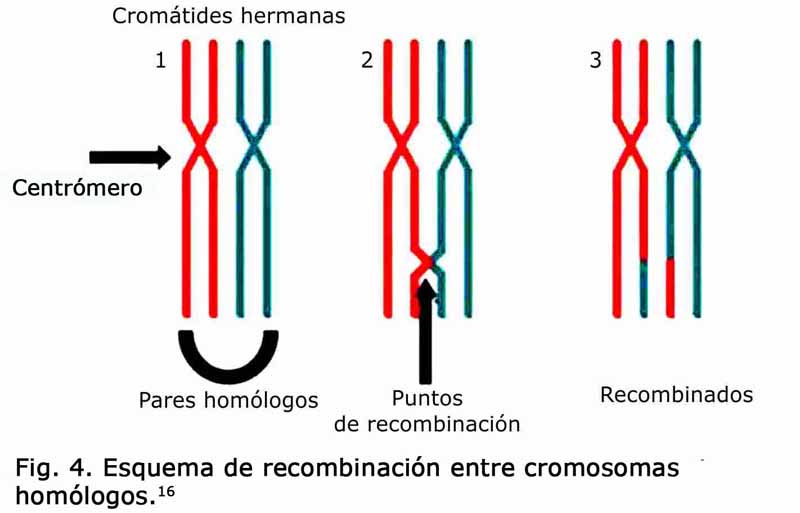
Estos rearreglos determinan la pérdida, ganancia o disrupción de genes cuya expresión fenotípica varía de acuerdo a la cantidad de secuencias codificantes presentes. Es importante comprender que para que este fenómeno, de rearreglo en la molécula de ADN por recombinación de cromosomas, ocurra debe existir como premisa la presencia de homología de sus secuencias aminoacídicas; este fenómeno se origina predominantemente en eventos de recombinación no alélica entre cromosomas homólogos (RNAH) (Fig. 4). Actualmente, y a diferencia de lo que antes se creía, los desórdenes genómicos y los eventos de recombinación no alélica entre cromosomas homólogos se consideran la principal causa de la mayoría de las enfermedades humanas.12,16,21
Comparativamente existen diferencias entre la mayoría de los trastornos de herencia monogénica (también llamados mendelianos) y los de causa genómica, en cuanto al origen y la etapa de producción de los mismos. Generalmente, los desórdenes monogénicos "convencionales" se deben a mutaciones específicas dentro de un gen y surgen por errores en la replicación o reparación del ADN. Los desórdenes genómicos, en cambio, se basan en un mecanismo de recombinación erróneo, que involucra a grandes segmentos de uno o más genes. 16,23 Debe destacarse además, que actualmente se ha descripto que varios trastornos mendelianos se originan también secundariamente a rearreglos cromosómicos.12
Se ha postulado que los rearreglos cromosómicos no ocurren al azar, sino que surgen en secuencias que presentan una estructura genómica compleja. Los puntos de quiebra implicados en estos rearreglos se distribuyen principalmente en regiones pericentroméricas y subteloméricas.8 Estos puntos de quiebra concentran particularmente regiones inestables de la molécula de ADN como lo son las secuencias repetidas llamadas LCRs (lowcopyrepeats) o los sitios palindrómicos ricos en adenina-timina.7
Para que un evento de recombinación se produzca es necesario que exista un segmento compartido de homología provisto por un sustrato. Las secuencias aminoacídicas repetidas (LCRs) situadas en cromosomas homólogos, actúan como sustratos de eventos de recombinación no alélica entre cromosomas homólogos (RNAH).7 La suma de estas secuencias particulares y los eventos RNAH son la fuente de origen de los principales rearreglos entre los que se incluye las deleciones (término técnico transliterado del inglés que significa amputaciones), las duplicaciones, las inversiones y las translocaciones.5 Los eventos RNAH que involucran LCRs que presentan una misma orientación, en el mismo cromosoma(ya sean intracromosómico o intracromátides), dan como resultado deleciones y/o duplicaciones. Los eventos RNAH ocasionados entre LCRs con orientación invertida, en el mismo cromosoma (ya sean intracromosómico o intracromátides), resultan en inversiones y aquellos que ocurren entre LCRs localizados en diferentes cromosomas dan como resultado translocaciones recíprocas 8,10,16 (Fig. 5).
ALGUNOS EJEMPLOS DE MALFORMACIONES CEFALOGÉNICAS SEGÚN EL NIVEL DE AFECTACIÓN EN LA CITODIFERENCIACIÓN
Diferentes alteraciones cefalogénicas tienen su origen en las alteraciones que generan la citodiferenciación en el nivel celular, molecular y en ocasiones en ambos niveles. A continuación señalaremos algunos ejemplos:
1) Ejemplos de rearreglos cromosómicos en la fisiopatología cefalogénica.
Dependiendo del tamaño del segmento genómico involucrado y de la cantidad de secuencia codificante presente en los genes contenidos en el segmento que sufre el rearreglo, pueden ocasionarse enfermedades mendelianas como la Neurofibromatosis de tipo 1 (NF1) de tipo autosómico dominante, causada por microdeleciones intersticiales en 17q11.2. Otro ejemplo es el síndrome de genes contiguos se origina en rearreglos del ADN (deleciones duplicaciones) que encierra varios genes adyacentes en un segmento del genoma. Entre ellos se incluye: el síndrome de Williams Beuren, los síndromes de PraderWilli-Angelman, la enfermedad de Charcot- Marie- Tooth tipo 1 y síndrome de deleción en 22q11.2. Existe una asociación entre la alteración del número de copias genómicas localizadas en el brazo largo del cromosoma 22 (22q) y la manifestación de varios síndromes entre los que se incluyen el síndrome de DiGeorge o velo-cardio-facial (22q11), caracterizado por ausencia del timo, de la glándula paratiroides, anomalías cardíacas, orejas de baja implantación, paladar fisurado y micrognatia; el síndrome de Pierre Robinyla fisura palatina aislada, entre otros.16,30-32
2) Ejemplos de la participación de los factores de crecimiento y sus receptores en la fisiopatología cefalogénica.
Los receptores de factor de crecimiento fibroblástico(FGFR) son moléculas de transducción de señales que atraviesan la membrana celular. Estos constituyen una familia de receptores tirosin-kinasa relacionados, pero individualmente diferentes que presentan 3 dominios similares a inmunoglobulina en la región extracelular (IgI, IgII, yIgIII), un dominio transmembrana, y un dominio citoplasmático tirosin-kinasa (TK1/TK2), que se trans-autofosforila. La gran mayoría de las mutaciones patogénicas en los FGFR son de cambio de sentido y confieren ganancia de función a la proteína mutada.16,21,33,34
En la fisiopatología de las craneosinostosis, de los síndromes de Pfeiffer, Jackson-Weiss,Antley-Bixler, la displasia osteoglofónica y el síndrome de Kallmann 2 autosómico dominante, juega un papel muy importante el receptor del factor de crecimiento fibroblástico-1(FGFR1).35-37 Este es codificado por 19exones, con una longitud del ARN transcripto de 4,095 bps y la longitud de la proteína es de 822 AA. Se encuentra localizado en 8p11.2-p11.1, este miembro de la familia FGFR está involucrado tanto para los factores de crecimiento fibroblástico básicos como ácidos y está implicado en la inducción de los primordios de los miembros inferiores. 3,5,11,38
Otro importante elemento en la citodiferenciación celular es el receptor del factor de crecimiento fibroblástico-2(FGFR2). Mutaciones en el FGFR2 se han descrito en las craniosinostosissindrómicasque aparecen en los síndromes de Apert, Crouzon y Pfeiffer. Estas mutaciones están abrumadoramente agrupadas en dos exones en FGFR2 llamados Hotspot, los cuales codifican para el dominio de la proteína IgIIIa/c8. Lo codifican 18 exones, con una longitud del ARN transcripto de 4,255 bps y una longitud de la proteína de 821 AA. Este miembro de la familia de los FGFR se encuentra localizado en 10q26, es un receptor de alta afinidad para los factores de crecimiento, ácidos, básicos yqueratinocitos, dependiendo de las isoformas correspondientes. Se han descrito variantes alternativas de empalme que codifican diferentes isoformas de proteínas, aunque no todas estas han sido completamente caracterizadas. 16,33,39
Por su parte, el receptor del factor de crecimiento fibroblástico-3 (FGFR3) está relacionado a las anomalías cefalogénicas que aparecen en la acondroplasia, la hipocondroplasia y el síndrome de Muenke, entre otros. Está codificado por 17 exones, con una longitud del ARN transcripto de 4,069 bps y la longitud de su proteína constituyente es de 806 AA. Este receptor del mencionado factor de crecimiento fibroblástico tiene una zona más proclive a las alteraciones genéticas denominada región flanqueante 5`que carece de caja:timina-adenina-timina-adenina (TATA) o citocina-adenina-adenina-timina (CAAT); sin embargo se han descrito diferentes sitios putativos para factores de transcripción como 0SP1 AP2, KROX24, IgHC.4,yZeste. Mutaciones en este gen localizadas en 4p16.3. se han visto fuertemente asociadas con la acondroplasia, la hipocondroplasia y el síndrome de Muenke. Dos mutaciones en el exon 10 resultan en una sustitución específica G380R responsable por el 97 % de los casos de acondroplasia.11,16,21,40
3) Ejemplo de alteraciones en el proceso de transcripción.
Un importante gen en la cefalogénesis lo constituye el denominado gen TWIST, pues es un factor de transcripción hélice-asa-hélice altamente conservado evolutivamente, lo que lo ha ubicado en la ejecución de funciones cruciales en el desarrollo embrionario y cefelogénicomesenquimal.La alteración de este gen se ha relacionado marcadamente a las acrocéfalo sindactilias, como el síndrome de SaethreChotzen. Este gen sido implicado en la diferenciación de las líneas celulares osteoblásticas. Se ha planteado que durante el desarrollo osteoblásticola presencia excesiva de la proteína que codifica el gen TWIST puede inhibir la maduración osteoblástica y mantener a estas células en un fenotipo preosteoblástico. Está determinado por un exón, con una longitud del ARN transcripto de 486 bps y una longitud protéica de 161 AA. Está localizado en locus 7p21.2. La patogenia causada se basa en el fenómeno de haploinsuficiencia, las mutaciones heterocigotas sin sentido (nonsense), con pérdida de función del gen. Este gen puede actuar como un inhibidor selectivo de la transcripción al reprimir la actividad de la acetil-transferasa de histonas.16,30,38,40
4) Ejemplo de alteraciones genéticas ligadas a la herencia.
Muchos son los novedosos descubrimientos que aportan evidencias de la participación genética ligada a la herencia en la fisiopatogenia de las fisuras labiales, alveolares y palatinas no sindrómicas (FLAPNS). Se ha demostrado que son varias las vías de producción de esta malformación cefalogénica, lo que además de su carácter no autosómico dominante, favorece frecuentemente el pronóstico de no aparición de dicha malformación en la descendencia de los portadores de FLAPNS.41,42 Se ha asociado su aparición a alteraciones epigenéticas en los cromosomas 4, 6 y X.3,16,43 Estudios realizados con marcadores microsatélites, en pacientes portadores de FLAPNS han demostrado la participación del loci 6p 22-25. En este cromosoma se ha descubierto la relación de la alteración de factores de crecimiento relacionada a esta malformación en el loci 6p, como lo es el caso de la familia del factor de crecimiento epidérmico (EGF), con mayor daño en la producción del factor de crecimiento transformador alfa (TGF-A).44La actividad de este loci ha sido demostrada además con el empleo de marcadores microsatélites MSX1.En la FLAPNS ha sido demostrada además la participación del locus 4q, a través del marcador microsatélite D4S192. Ha sido también demostrada la participación del cromosoma X a nivel de sus lociXq 21.1 y Xq 21.31.6,16,45,46
Estas alteraciones genéticas, ligadas mayormente a la herencia, condicionan la aparición de alteraciones moleculares similares en casi todos los casos de FLAPNS, como son: la disminución del nivel de glicoproteínas en hueso palatino, la alteración de la actividad del factor-6 regulador de interferón (IRF6), la inactivación del receptor tipo 1 para las proteínas morfogénicas óseas BMP (Bmpr1a) en el hueso palatino, lo que genera disminución del tejido mesenquimal maxilar y la inactivación del gen Bmp4 que determina la BMP tipo 4,lo cual genera la aparición de fisura labial aislada y de alteraciones en la dentinogénesis de la zona de la fisura.16,22,43,45
Otras consecuencias moleculares de las alteraciones epigenéticas en la FLAPNS son, la disminución del nivel del factor de crecimiento embrionario (EGF) y del factor de crecimiento tumoral beta 1 (TGF â1) que están involucrados en la síntesis de ADN en el mesénquima; así como la alteración de las concentraciones del adenosínmonofosfato-quinasa (AMP-quinasa) y del EGF que intervienen en la formación del paladar secundario. Aparece además la disminución del ácido retinoico que participa en la arquitectura del mesénquima palatino, así como de las metaloproteinasas de la matriz (MMP) que también intervienen en la morfogénesis palatina en relación a la actividad del TGF â3.16,44,45
TEORÍAS DE LAS MALFORMACIONES CEFALOGÉNICAS
La filosofía positivista que invadió a las universidades de continente americano a finales del siglo XIX y principios del siglo XX, trajo consigo un deterioro del análisis epistemológico de las ciencias. Esta concepción en la formación curricular de las universidades generó un efecto reduccionista de las ciencias médicas y estomatológicas, pues la formación académica se concentró en aportar lo esencial para el ejercicio de la profesión sin potencializar una amplia incorporación de principios científicos y metodológicos investigativos que favorecieran el desarrollo teórico-conceptual y científico-técnico en la búsqueda de la evidencia científica. El estigma del academicismo disciplinar generó que las especialidades crearan "feudos" en el conocimiento científico, lo cual impidió la interdisciplinariedad y la transdisciplinariedad en la solución de los problemas de la profesión; la participación colaborativa de las diferentes especialidades era puntual y formal, no se generaban aportes teórico-conceptuales sólidos.47
Consecuentemente, la filosofía que en muchas latitudes imperó fue la dialéctica hegeliana, atemperada a diferentes escuelas de pensamiento como el utilitarismo y el materialismo, entre otras. Esta concepción filosófica presuponía una visión pragmático-utilitarista de los fenómenos que se estudiaban, pues los simplificaba a un nivel bicategorial de unidad y lucha de contrarios, donde una categoría filosófica se contraponía a la otra. Hoy conocemos que los fenómenos sociales, psicológicos y biológicos requieren para su comprensión más que un análisis de dos categorías contrapuestas y el materialismo histórico-dialéctico ha abrazado en la actualidad la teoría de los sistemas complejos.14,15 Los análisis matemáticos que determinaron los elementos fractales y el análisis no lineal de los fenómenos incluso biomédicos, determinó un nuevo sendero para la comprensión de los procesos que integran sistemas mucho más complejos de lo que el hombre imaginó, donde la explicación es solo posible a través de la construcción de metaestructuras o redes organizacionales en distintos dominios que se imbrican a través del análisis de atractores de clausura o apertura disciplinar, díganse: conceptos, teorías, métodos de investigación e interacción interdisciplinar.48,49
En este sentido, la reflexión epistemológica de la prospectiva universitaria en la formación estomatológica abre las fronteras entre todas las disciplinas afines naturales y humanísticas, lo que permite generar convergencias en vez de trazar límites demarcatorios. La reflexión no es patrimonio de ningún dominio disciplinar, es la actitud más rica del conocimiento, el momento en que este es capaz de autoconsiderarse y de metasistematizarse. Las posibilidades de conceptualizar una teoría del conocimiento para las ciencias estomatológicas es hoy una tarea capital a la vez de aleatoria y compleja; para ello hay que comprender que la revolución científico-técnica que necesita la estomatología en nuestro país y en Latinoamérica debe ser desarrollada hoy, no en la oposición bicategorial simplista, sino en el terreno de la complejidad transdisciplinar del modo de organización teórico-conceptual del conocimiento, lo que exige pensar la instrumentación teórico-conceptual estomatológica de forma radicalmente compleja. A partir de ese enfoque se construirá un nuevo paradigma pedagógico y científico de la complejidad transdisciplinar como una necesidad epistemológica fundamental para la estomatología.
Por eso, hoy es posible evaluar de manera más integradora la cefalogénesis, no como un área del conocimiento de las ciencias básicas, sino como una materia de obligada pertinencia para todas las especialidades médicas y estomatológicas que tienen como campo de acción profesional el estudio de la morfofisiología y las enfermedades de la cabeza y el cuello, así como su tratamiento médico-quirúrgico y estético-rehabilitador.50
Históricamente la explicación de las malformaciones craneofaciales han estado basadas en lo que el autor de este artículo ha denominado teorías monotisulares-determinantes, pues involucran a un solo tejido cefalogénico y la supuesta alteración está determinada a nivel de un tejido específico sin la participación de otros niveles o estructuras morfogénicas. Desde finales del siglo XIX existieron dos teorías mayormente conocidas, la teoría que plantea la aparición de las malformaciones craneofaciales por «un fallo en la fusión» y la teoría que plantea que estas ocurren por un «retraso en la migración mesodérmica», con diferentes aportes, pero sobre el presupuesto teórico de una u otra teoría. Posteriormente Van der Muelen en el siglo XX propuso una teoría más compleja basada en los eventos embriológicos.51 A continuación las describiremos.
1) Teoría del fallo de la fusión de Dursy y His: esta teoría fue propuesta por los doctores Dursy y His en el siglo XIX y es considerada la teoría clásica y habitualmente se excluye a uno de sus autores, por lo que es mayormente conocida como la teoría de His. Esta teoría propone que las malformaciones cefalogénicas ocurren por un fallo en la fusión de los diferentes procesos o en algún punto particular de esos procesos, lo que produce un retardo o enlentecimiento en su crecimiento lo que genera una ausencia de la fusión con otros procesos y consecuentemente aparece la fisura o defecto craneofacial.3,6,51,52
2) Teoría de la persistencia de las células epiteliales de Warbrick: básicamente Warbrickse acogió a la teoría de Dursy y His, pero la modificó al argumentar que la causa de la ocurrencia del fallo de la fusión planteada en la teoría clásica se debía a la persistencia de las células epiteliales que normalmente debían desaparecer para que ocurriera la fusión de los procesos, por lo que si la cubierta epitelial persistía el mesodermo de un proceso no podía fusionarse con el mesodermo opuesto, lo cual generaría una solución de continuidad en esa región.3,6,51,52
3) Teoría de la migración mesodérmica de Pohlmann y Veau: esta teoría fue presentada por PohlmannyVeaua inicios del siglo XX, ellos explicaron que las alteraciones en la cefalogénisisocurrían por la ausencia de migración y penetración mesodérmica lo que generaba el colapso del ectodermo causado por la ausencia de soporte mesodérmico, este colapso es el que finalmente produce la fisura.3,6,51
4) Teoría de Van der Muelen: en la segunda mitad del siglo XX Van der Muelen y otros propusieron una teoría más compleja en la que sus conceptos embriológicos están mayormente relacionados con la aparición de las anomalías por fisuras. El planteó que las malformaciones fisurales no son fisuras realmente, sino displasias. Estas displasias son el resultado del retardo del desarrollo durante la fusión de los procesos faciales. El defecto resultante es causado por la ausencia o el insuficiente desarrollo de los centros de osificación.3,6,51
5) Teoría de McKenzie y Craig: ante la evidencia clínica de la aparición de ciertas fisuras que no seguían una línea de fusión específica de los procesos embrionarios faciales o craneales McKenzie y Craig propusieron en el año 1955 su teoría que con la anteriormente mencionada justificación de casos clínicos plantearon que las malformaciones craneofaciales ocurrían debido a un fallo a nivel de la cresta neural en cualquier momento de la migración celular o por fallo vascular inducida por la atrofia de los vasos sanguíneos. Esta teoría tomó mucho auge pues en el 1975 Poswillo demostró que el síndrome del primer y segundo arcos branquiales podía ser reproducido en el laboratorio cuando ocurre la coaptación de la arteria estapédica.3,6,51
PROPUESTA DE UNA TEORÍA INTEGRADORA
Históricamente se trató de explicar los complejos procesos biológicos cefalogénicos a través de una concepción reduccionista que con teorías no demostradas planteaba como mecanismo de aparición de las malformaciones cráneo-maxilofaciales a los efectos secundarios ocurridos en los tejidos sin comprender sus liados mecanismos celulares y moleculares. Estas teorías no explican las esencias causales de las alteraciones cefalogénicas y nos conducen a realizar una recurrente pregunta que no tuvo respuesta durante muchas décadas: ¿por qué ocurren estas fallas de proliferación mesodérmica o persistencia del tejido ectodérmico?
Resulta evidente que todas las teorías antes mencionadas aluden a consecuencias de una alteración primaria que no se explica totalmente al describir lo que sucede en el nivel de organización tisular de la cefalogénesis. Debe entonces recurrirse al nivel celular y molecular para su comprensión, debe entenderse la epigenética, la participación de los homeogenes, de los factores de crecimiento de la transcripción del ADN y de este al ARN hasta la síntesis de proteínas específicas que condicionan la citodiferenciación celular de la propia célula o de otras que se encuentran distantes. En medio de todo esto es necesario comprender la importancia de la cronometría del ADN nuclear, donde los segmentos cromosómicos leaders están predeterminados para la activación del segmento capy que condicionan la inducción de una nueva síntesis proteica en un momento preciso del desarrollo cefalogénico. Importante papel juega el medio ambiente con el aporte de factores teratogénicos que pueden afectar el normal desenvolvimiento cefalogénico, así como estados carenciales de la madre, la presencia de hábitos tóxicos, entre otros que condicionan un complejo fenómenos probabilístico donde hasta el azar puede influir. No debe olvidarse el ADN mitocondrial que interviene también en los procesos citoplasmáticos citoinductores, lo cual no depende del ADN nuclear.
Es perentorio entonces, que no se trate de explicar las alteraciones de la cefalogénesis desde concepciones reduccionitas como las ya obsoletas teorías de Dursy y His o la de Pohlmann y Veau, o pretender categorizar los porcentajes de influencias del medio ambiente o de la genética, los cuales son útiles en la genética clínica, pero no explican el proceso pretendido. La cefalogénesis debe ser comprendida como un fenómeno mucho más complejo, pero sujeto a principios esenciales. Es por eso que proponemos en este trabajo una teoría holístico-sinérgica de la cefalogénesis, que explica a la cefalogénesis como el resultado de la conjugación de complejos factores: hereditarios, citoplasmáticos y medioambientales que dan lugar a una morfología determinada, genética y molecularmente, por la citoinducción y la citodiferenciación de los tejidos participantes en su formación.
Por todo lo antes expuesto y como resultado de esta investigación se propone, a la comunidad científica que tiene como objeto de estudio y praxis médica la región craneofacial, la teoría holístico-sinérgica de las malformaciones cefalogénicas, la cual considera a las malformaciones cefalogénicas como el complejo resultado de la alteración en la transcripción genética y/o en la síntesis molecular de proteínas específicas que determinan una citoinducción o una citodiferenciación anormal por diversas causas: hereditarias, citoplasmáticas o medioambientales, las cuales pueden actuar de maneras aisladas o superpuestas y que generan una dismorfiacraneofacial con secuelas estructurales (morfológicas), fisionómicas (estéticas), y quinésicas (funcionales) que diferencian a su portador de los estándares fisiológicos establecidos en los humanos.
DISECCIÓN ETIMOLÓGICA
Se denominó como holísticas tanto a la teoría de la cefalogénesis, como a la teoría de las malformaciones congénitas por cuanto en la teoría filosóficade los sistemas el holismo es la concepción y presupuesto teórico de que las propiedades de un sistema no pueden determinarse con la simple suma de sus partes o analizando sus partes de forma individual, sino que las partes o componentes deben verse como un todo. En este sentido se basa el holismo en la concepción de Aristóteles que se resume en su frase: "el todo es más importante que la suma de sus partes". Esto significa que no se trata de plantear esquemáticamente que en la cefalogénesis fisiológica o patológica intervienen diferentes factores, pues hay que considerar la conjugación de todos ellos en las esencias integradoras de sus procesos, donde no se deben establecer proporciones categóricas por ser la cefalogénesis el resultado de un entramado mecanismo y de reacciones complejas.47
Esta concepción se aviene a la teoría filosófica de los sistemas complejos, donde cada elemento desempeña un importante papel y en cada ocasión pueden variar sus proporciones de interacción.47 Consecuentemente, la teoría de los sistemas complejos aplicada a la cefalogénesis la despoja del reduccionismo pragmático utilitarista que imperó en la medicina el siglo XX, que pretendió deducir todas las propiedades de un sistema mediante el mero análisis de sus componentes. Nadie puede categorizar qué acción determinante va a tener en una cefalogénesis específica la predisposición genética familiar, o la acción de un determinado agente externo con potencial teratogénico o el estado nutricional o endocrino-metabólico de la madre; como tampoco se pueden categorizar qué mecanismos compensadores y rectificadores de la genética y la citodiferenciación celular anómalas intervendrán en la reducción dela penetrancia o expresividad fenotípica de una determinada malformación cefalogénica.
Por ejemplo: si la falta de un aminoácido esencial secundario a un estatus carencial generó en el citoplasma embrionario una alteración en la síntesis proteica específica de un polipéptidocitoinductor a distancia, lo que redundaría en un fallo de la citodiferenciación de un tejido determinado en la cefelogénesis, el sistema de cremallera Poli I y Poli II que rectifica la secuencia aminoacídica luego de la síntesis proteica puede detectar la falla y consecuentemente rectificarlo, a través de sistemas de señales moleculares intracitoplasmáticos o extracitoplasmáticos para la obtención del aminoácido esencial y su rectificación, para bloquear así el efecto nocivo del factor teratogénico. Asimismo, lo que es probable matemática y epidemiológicamente, puede no ocurrir por factores emergentes o poco relevantes no analizados en las ecuaciones bioestadísticas.
La terminología sinérgica fue empleada por el autor para calificar a las teorías presentadas, por cuantoen la teoría filosófica de los sistemas la sinergia hace referencia al efecto interactivo de las partes del sistema, donde la suma de la interacción de las partes componentes de la organización es mayor que el efecto de las partes por separado. Expresado de otro modo y llevado a una concepción matemática, el sinergismo genera resultados exponenciales donde «2+2= 5».47 Esto se explica comprendiendo que la superposición de factores aleatorios participantes en la cefalogénesis puede potenciar un efecto negativo o positivo.
Por citar un ejemplo hipotético, la presencia de una predisposición genética a desarrollar una craneosisnostosis no sindrómica de tipo recesiva sin expresividad fenotípica en la ascendencia del feto, puede potenciarse sinérgicamente por la ingestión mantenida de yogurt de soya por la gestante durante todo el embarazo. La soya contiene un alto porcentaje de vitamina A, la cual tiende a favorecer la aparición de craneosinostosis. Bajo esta condición nutricionalel feto que no iba a desarrollar una malformación craneal sí la desarrolla por el sinergismo de un factor sobre otro. Si además aparece un hábito tóxico de drogadicción que interfiere en la correcta síntesis proteica fetal se potenciará el agravamiento de esta malformación craneal que en otras condiciones no hubiera aparecido.
La teoría de los sistemas complejos incluye el análisis de los fenómenos biomédicos a través de la comprensión de metaestructuras o de redes funcionales en distintos dominios o niveles de organización de los sistemas (molecular, celular, tisular) relacionadas entre sí, holística y sinérgicamente. En tal sentido la cefalogénesis fisiológica o patológica debe ser entendida de manera holística por cuanto es un complejo sistema que debe considerarse como un todo. Los cambios de cualquier parte de este complejo sistema morfogénico tienen impacto y le afectan en su totalidad. De igual manera la cefalogénesis fisiológica o patológica debe ser entendida de manera sinérgica pues como resultado de esas interacciones entre los elementos participantes, surgen propiedades o cualidades mofológicas nuevas o emergentes, que no pueden explicarse analizando sus elementos constituyentes de forma aislada. Por esta razón, mientras más complejo sea un sistema, mayor serán sus probabilidades de variaciones cualitativas o funcionales.
CONCLUSIÓN
Es pertinente actualizar la manera en que se estudia y se enseña la cefalogénesis y sus alteraciones en nuestro medio, para proveer una concepción más integradora y científica, desde el conocimiento de los diferentes niveles biológicos que participan en ella. Sugerimos que se implementen en los planes de estudio de pregrado en la carrera de Estomatología y de posgrado la teoría holístico-sinérgica de la cefalogénesis y sus malformaciones, donde la complejidad de interacciones genéticas, ambientales y citoplasmáticas condicionan la eugenesis o la presencia de alteraciones epigenéticas o citoplasmáticas que determinan la aparición de citodiferenciación anómala de uno o más tejidos embrionarios o fetales, con repercusión fenotípica morfofuncional.
REFERENCIAS BIBLIOGRÁFICAS
1. Vila Morales D. Teoría del Derecho Médico. La Habana: Editorial Ciencias Médicas; 2012.
2. Lonai P, Orr-Urtreger A. Homeogenes in mammalian development and the evolution of the cranium and central nervous system. FASEB J. 1990;4:1436-43.
3. Vila Morales D. Actualización en las malformaciones congénitas craneofaciales. Conferencia de la Jornada Científica de Cirugía Maxilofacial "Ana Larralde" In Memoriam; 2012 May 3-4; La Habana, Cuba.
4. Vila Morales D. Presentación de una nueva clasificación integradora de las malformaciones craneofaciales. Rev Habanera de Ciencias Médicas. 2006[citado: 12 May 2012];5(3): [aprox. 10 p.]. Disponible en: http://www.ucmh.sld.cu/rhab/rhcmv5n3.htm.
5. Vila Morales D. Alteraciones del desarrollo del cráneo, la cara, la boca y el cuello En: Santana Garay JC. Atlas de patología del complejo bucal. La Habana: Editorial Ciencias
Médicas; 2010[citado: 15 May 2012]. Disponible en: http://www.bvs.sld.cu/libros/atla_cancerbuc/indice_p.htm
6. Sperber GH. Craniofacial embriology. Oxford: Oxford University Press; 1994.
7. Sperber GH, Machin AG. The enigma of cephalogenesis. Cleft Palate-Craniofacial Journal. 1994;31(2):91-6.
8. Shaw JC, Lupski JR. Implications of human genome architecture for rearrangements-based disorders: the genomic basis of disease. Human Molecular Genetics. 2004;13:57-64.
9. Sassoon DA. Homeogenes and vertebrate skeletal miogenesis: a perspective. Human Molecular Genetics. 1996;6(6):431-6.
10. Kumar D. Disorders of the genome architecture: a review. Genomic Medicine. 2008;2(3):69-76.
11. Cole P, Kaufman Y, Hatef DA, Hollierc LH. Embryology of the Hand and Upper Extremity. Journal of Craniofacial Surgery. 2009;20(4):992-5. doi:10.1097/SCS.0b013e3181abb18e
12. Lupski JR and Stankiewicz P. Genomic disorders: Molecular Mechanisms for Rearrangements and Conveyed Phenotypes. Plos Genetics. 2005;1(6):627-33.
13. Santiago P, TremblayK, BasriE E, Arnal E. Tertiary Education for the Knowledge Society. Thematic Review of Tertiary Education Journal. 2008;2(3):321-4.
14. Austin W, Park C, Goble E. From interdisciplinary to transdisciplinary research: A case study. Qualitative Health Research. 2008;18:557-64.
15. Cambursano S. Interdisciplina, transdisciplina y multidisciplina. Prácticas en docencia e investigación. [Doctorado en Ciencias Humanas]. Facultad de Humanidades. Universidad Nacional de Catamarca, Argentina; 2006.
16. Ramírez JM, Echeverría MI, Mampel A, Marino M, Gallardo A, Triguy J, et al. MLPA en el estudio de desórdenes genómicos. Revista Médica Universitaria-Argentina. 2009;5(2):143-6.
17. Choi JW, Park HW, Kwon YJ, Park BY. Role of Apoptosis in Retinoic Acid-Induced Cleft Palate. Journal of Craniofacial Surgery. 2011;22(5):1567-71. doi:10.1097/SCS.0b013e318208ba10.
18. Captier G, Dessauge D, Picot MC, Bigorre M, Gossard C, El Ammar J, Leboucq N. Classification and Pathogenic Models of Unintentional Postural Cranial Deformities in Infants: Plagiocephalies and Brachycephalies. Journal of Craniofacial Surgery. 2011;22(1):33-41. doi:10.1097/SCS.0b013e3181f6c386.
19. Ridgway EB, Wu JK, Sullivan SR, Vasudavan S, Padwa BL, Rogers GF, et al. Craniofacial Growth in Patients With FGFR3Pro250Arg Mutation After Fronto-Orbital Advancement in Infancy. Journal of Craniofacial Surgery. 2011;22(2):455-61. doi:10.1097/SCS.0b013e3182077d93.
20. Smith DM, Cooper GM, Mooney MP, Marra KG, Losee JE. Bone Morphogenetic Protein 2 Therapy for Craniofacial Surgery. Journal of Craniofacial Surgery. 2008;19(5):1244-59. doi:10.1097/SCS.0b013e3181843312.
21. Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends in Genetics. 2002;18(2):74-82.
22. Vila Morales D, Leyva Mastrapa T, Alonso FernándezL, Sánchez Cabrales E, Lazo Montero JC. Equipo cubano interdisciplinario de cirugía craneofacial en pediatría. Resultados de un quinquenio. Rev Cubana Estomatol. 2010[citado: 12 May 2012];47(1):[aprox. 15 p.]. Disponible en: http://www.bvs.sld.cu/revistas/est/vol_47_01_10/est06110.htm
23. Laroche F, Ramoz N, Leroy S, Fortin C, Rousselot-Paillet B, Philippe A, et al. Polymorphisms of coding trinucleotide repeats of homeogenes in neurodevelopmental psychiatric disorders.Psychiatr Genet. 2008;18(6):295-301.
24. Mansfield JH, McGlinn E. Evolution, expression, and developmental function of Hox-embedded miRNAs. Curr Top Dev Biol. 2012;99:31-57.
25. Oulion S, Laurenti P, CasaneD.Hox genes organization: studying non-model vertebrates leads to a paradigm shift. Med Sci (Paris). 2012;28(4):350-3.
26. Paina S, Garzotto D, DeMarchis S, Marino M, Moiana A, Conti L, et al. Wnt5a is a transcriptional target of Dlxhomeogenes and promotes differentiation of interneuron progenitors in vitro and in vivo. J Neurosci. 2011;31(7):2675-87.
27. Pick L, Heffer A. Hox gene evolution: multiple mechanisms contributing to evolutionary novelties.Ann N Y Acad Sci. 2012;12(56):15-32. doi:10.1111/j.1749-6632.2011.06385.x.
28. Rajshankar D, Downey GP, McCulloch CA. IL-1â enhances cell adhesion to degraded fibronectin. FASEB J. 2012;21(4):234-9.
29. Liu F, van der Lijn F, Schurmann C, Zhu G, Chakravarty MM, et al. A Genome-Wide Association Study Identifies Five Loci Influencing Facial Morphology in Europeans. PLoSGenet. 2012;8(9):45-9.
30. Sepúlveda GT, Palomino ZH, Cortés AJ. Prevalencia de fisura labiopalatina e indicadores de riesgo: Estudio de la población atendida en el Hospital Clínico Félix 31. Bulnes de Santiago de Chile. RevEspCir Oral y Maxilofac. 2008;30(1):17-25.
31. Mackay DR. Controversies in the Diagnosis and Management of the Robin Sequence. Journal of Craniofacial Surgery. 2011.22(2):415-20. doi:10.1097/SCS.0b013e3182074799.
32. Wojciech D, Warren SM. Current Concepts in Deformational Plagiocephaly. Journal of Craniofacial Surgery. 2011;22(1):6-8. doi:10.1097/SCS.0b013e3182074e04.
33. van der Meulen J, van der Hulst R, van Adrichem L, Arnaud E, Chin-Shong D, Duncan C, et al. The Increase of Metopic Synostosis: A Pan-European Observation. Journal of Craniofacial Surgery. 2009;20(2):283-6. doi:10.1097/SCS.0b013e31818436be
34. Marchac A, Arnaud E, Di Rocco F, Michienzi J, Renier D. Severe Deformational Plagiocephaly: Long-Term Results of Surgical Treatment. Journal of Craniofacial Surgery. 2011;22(1):24-9. doi:10.1097/SCS.0b013e3181f7dd4a
35. Levi B, Wan DC, Longaker MT, Habal MB. Deformational Plagiocephaly: A Look Into the Future. Journal of Craniofacial Surgery. 2011;22(1):3-5. doi:10.1097/SCS.0b013e3181fb7ee5
36. Kolar JC. An Epidemiological Study of Nonsyndromal Craneosinostosis. Journal of Craniofacial Surgery. 2011;22(1):47-9. doi:10.1097/SCS.0b013e3181f6c2fb.
37. Kandasamy J, Anderson K, Dunne J, Grogan J, Duncan C, Sinha A, May P. Treatment of ScaphocephalyWith Combined Vertex Craniectomy and Bilateral Microbarrel Staving. Journal of Craniofacial Surgery. 2011;22(1):42-6.
38. Rogers, GF. Deformational Plagiocephaly, Brachycephaly, and Scaphocephaly. Part I: Terminology, Diagnosis, and Etiopathogenesis. Journal of Craniofacial Surgery. 2011; 22(1):9-16. doi:10.1097/SCS.0b013e3181f6c313.
39. Rogers GF. Deformational Plagiocephaly, Brachycephaly, and Scaphocephaly. Part II: Prevention and Treatment. Journal of Craniofacial Surgery. 2011;22(1):17-23. doi:10.1097/SCS.0b013e3181f6c342
40. Domeshek LF, Das RR, Van Aalst JA, Mukundan SJ, Marcus JR. Influence of Metopic Suture Fusion Associated With Sagittal Synostosis. Journal of Craniofacial Surgery. 2011;22(1):77-83. doi:10.1097/SCS.0b013e3181f6c56b.
41. Dudas JR, Losee JE, Penascino VM, Smith DM, Cooper GM, Mooney PM, et al. Leporine-Derived Adipose Precursor Cells Exhibit In Vitro Osteogenic Potential. Journal of Craniofacial Surgery. 2008;19(2):360-8. doi:10.1097/SCS.0b013e318163e17b.
42. Ichida M, Komuro Y, Yanai A. Consideration of Median Cleft Lip With Frenulum Labii Superior. Journal of Craniofacial Surgery. 2009;20,5:1370-4. doi:10.1097/SCS.0b013e3181ae42bf.
43. Kim SM, Lee JH, Jabaiti S, Lee SK, Choi JY. Tbx22 Expressions During Palatal Development in Fetuses With Glucocorticoid-/Alcohol-Induced C57BL/6N Cleft Palates. Journal of Craniofacial Surgery. 2009;20(5):1316-26. doi:10.1097/SCS.0b013e3181ae6686.
44. Kramer FJ, Gruber R, Fialka F, Sinikovic B, Schliephake H. Quality of Life and Family Functioning in Children With Nonsyndromic Orofacial Clefts at Preschool Ages. Journal of Craniofacial Surgery. 2008;19(3):580-7. doi:10.1097/SCS.0b013e31816aaa43.
45. Manna F, Pensiero S, Clarich G, Guarneri GF, Parodi PC. Cleft Lip and Palate: Current Status From the Literature and Our Experience. Journal of Craniofacial Surgery. 2009;20(5):1383-7. doi:10.1097/SCS.0b013e3181b0daa3.
46. Stoler JM, Rosen H, Desai U, Mulliken JB, Meara JG, Rogers GF. Cleft Palate in Pfeiffer Syndrome. Journal of Craniofacial Surgery. 2009;20(5):1375-7. doi:10.1097/SCS.0b013e3181ae42e4.
47. Vila Morales D. Aplicación de distracción en hipoplasias mandibulares. Propuesta de un modelo antropométrico para su evaluación. Tesis de Doctor en Ciencias Médicas. La Habana: Universidad de Ciencias Médicas de La Habana; 2005.
48. Espina, M. Complejidad, transdisciplina y metodología de la investigación social. Utopía y praxis latinoamericana. Redalyc UAEM.2007;12(38):29-43.
49. GreckhamerT, Kro-Ljungberg M, Cilesiz S, Hayes S. Demystifying Interdisciplinary. Qualitative Research Qualitative Inquiry. 2008;14(2):307-31.
50. Vila Morales D, Leyva Mastrapa T, Alonso Fernández L. Aportes y modificaciones de técnicas quirúrgicas en cirugía craneofacial pediátrica. Rev Cubana Estomatol. 2010[citado: 10 May 2012];47(3):[aprox. 11 p.]. Disponible en: http://bvs.sld.cu/revistas/est/vol_47_03_10/est04310.htm
51. RareCranio-Facial Clefts. In: Plastic and ReconstructiveSurgery. Cirujanos PlástiKosMundi. Bogotá: Editorial Alaire. [En línea]. 2012. [citado: 10 julio 2012]. Disponible en: http://www.cpmundi.org/adjuntos/manuales/es/rare_cranio-facial_clefts -5.pdf
52. Kruger GO. Textbook of Oral and Maxillofacial Surgery. St Louis, USA: Mosby Co.; 1979. p. 98-106.
Recibido: 14 de mayo de 2012.
Aprobado: 6 de julio de 2012.
Dr. CM. Dadonim Vila Morales. Facultad de Ciencias Médicas "Finlay-Albarrán". Universidad de Ciencias Médicas de La Habana, Cuba. Correo electrónico: dadonim.vila@infomed.sld.cu

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}