










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Medicina
versión On-line ISSN 1561-302X
Rev cubana med v.47 n.1 Ciudad de la Habana ene.-mar. 2008
PRESENTACIÓN DE CASOS
Colangitis esclerosante primaria en paciente con panhipopituitarismo autoinmune. Presentación de 1 caso
Primary sclerosing cholangitis in a patient with autoimmune panhypopituitarism. A case report
Rolando Rodríguez FernándezI; Emilio Buchaca FaxasII; Nelson Roselló SilvaIII; Lays Rodríguez AmadorIV; Miguel Ángel YánezI; Carlos Domínguez ÁlvarezV
IEspecialista de I Grado en Medicina Interna. Instructor. Hospital Clinicoquirúrgico "Hermanos Ameijeiras". La Habana, Cuba.
II Doctor en Ciencias Médicas. Especialista de II Grado en Medicina Interna. Profesor Auxiliar. Hospital Clinicoquirúrgico "Hermanos Ameijeiras". La Habana, Cuba.
III Especialista de II Grado en Medicina Interna. Profesor Auxiliar. Hospital Clinicoquirúrgico "Hermanos Ameijeiras". La Habana, Cuba.
IV Especialista de I Grado en Medicina Interna. Profesora Auxiliar. Hospital Clinicoquirúrgico "Hermanos Ameijeiras". La Habana, Cuba.
V Especialista de II Grado en Anatomía Patológica. Profesor Auxiliar. Hospital Clinicoquirúrgico "Hermanos Ameijeiras". La Habana, Cuba.
RESUMEN
Se presentó un paciente con diagnóstico de un panhipopituitarismo autoinmune (diabetes insípida vasopresinsensible, hiperprolactinemia, hipotiroidismo, hipogonadismo secundario e insuficiencia corticosuprarrenal) establecido hace 3 años, que a finales del año 2005 comenzó a presentar dolor intenso en epigastrio e hipocondrio derecho, acompañado de vómitos con restos de alimentos, fiebre elevada con escalofríos, seguido de coluria, acolia e íctero verdínico, hepatomegalia, elevación importante de la bilirrubina, la fosfatasa alcalina leucocitaria y la gamma glutamil transpectidasa, leucocitosis, que requirió tratamiento con antibióticos por vía endovenosa y simuló, al inicio, los diagnósticos de litiasis coledociana, tumor de cabeza de páncreas y hepatitis colangiolítica por fármacos. Estos episodios se fueron repitiendo cada vez con más frecuencia, hasta llegarse al diagnóstico de colangitis esclerosante primaria por colangiopancreatografía retrógrada endoscópica y biopsia hepática. Se revisó la literatura, pero no se encontraron reportes de la asociación de colangitis esclerosante primaria con panhipopituitarismo autoinmune
Palabras clave: Colangitis esclerosante primaria, panhipopitiutarismo autoinmune, transplante hepático.
ABSTRACT
A case of a patient with diagnosis of autoimmune panhypopituitarism (vasopressinsensitivity diabetes insipidus, hyperprolactinemia, hypothyroidism, secondary hypogonadism and corticosuprarenal insufficiency) established 3 years ago, was reported. At the end of 2005, he began to present an acute pain in the epigastrium and right hypochondrium, accompanied with vomiting with food residual, high fever with chills, followed of choluria, acholia and greenish icterus, hepatomegaly, marked elevation of bilirubin, leukocyte alkaline phosphatase and gamma glutamil transpeptidase, leukocytosis, that required treatment with antibiotics by endovenous route and mimmicked, at the beginning, the diagnoses of choledocian lithiasis, pancreatic head tumor, and cholangiolitic hepatitis caused by drugs. These episodes were frequently repeated until the diagnosis of primary sclerosing cholangitis was made by retrograde endoscopic cholangiopancreatography and liver biopsy. Literature was reviewed, but no reports of association of primary sclerosing cholangitis with autoimmune panhypopituitarism were found.
Key words: Primary sclerosing cholangitis, autoimmune panhypopitiutarism, liver transplant.
La colangitis esclerosante primaria (CEP) es una enfermedad de la vía biliar, tanto intrahepática como extrahepática, caracterizada por inflamación crónica seguida de obliteración fibrosa de la luz que ocasiona colestasis obstructiva.1-3 La CEP es la más común de todos los tipos de colangitis esclerosante, es la cuarta causa de trasplante hepático en los EE. UU. y equivale al 1 % de todos los trastornos hepáticos. Se estima que su prevalencia se sitúa entre 1 y 5 casos por 100 000 hab.4 Puede considerarse una enfermedad preneoplásica, ya que aproximadamente el 10 % de los casos desarrolla un colangiocarcinoma.5,6 Se ha descrito la asociación con diversas enfermedades, como: colitis ulcerosa y enfermedad de Crohn7-9 y, menos frecuente, con otras como la enfermedad celíaca, diabetes mellitus, artritis reumatoidea, síndrome de Sjögren´s, esclerosis múltiple, fibrosis retroperitoneal, pancreatitis crónica, sarcoidosis, tiroiditis, nefritis lúpica, anemia autoinmune, púrpura trombocitopénica idiopática, vasculitis, litiasis biliar10-12 y enfermedades neoplásicas como el colangiocarcinoma, el cáncer de vesícula, hígado y de colon. Otras enfermedades descritas con menor frecuencia asociadas a la CEP son la hepatitis autoinmune y la cirrosis biliar primaria.13,14
La asociación de esta enfermedad con el panhipopituitarismo primario autoinmune no la encontramos reportada en la literatura.
La causa de la CEP todavía se desconoce. Se ha sugerido una etiología infecciosa, vírica o bacteriana, o que se trate de una enfermedad autoinmune que se desarrollaría en personas genéticamente predispuestas .El diagnóstico suele basarse en una combinación de antecedentes clínicos, alteraciones bioquímicas, radiológicas e histológicas.15-17
PRESENTACIÓN DEL CASO
Paciente masculino de 53 años de edad, raza blanca, con antecedentes diagnósticos de una angina vasoespástica diagnosticada hace 5 años, y de un panhipopituitarismo autoinmune (diabetes insípida, vasopresinsensible, hiperprolactinemia, hipotiroidismo, hipogonadismo secundario e insuficiencia corticosuprarrenal secundaria) para el que se le administró tratamiento sustitutivo con hormonas y presentó mejoría clínica y humoral, aunque refiere episodios de recaídas.
A finales del año 2005 comenzó a presentar dolor intenso en epigastrio e hipocondrio derecho acompañado de vómitos con restos de alimentos, fiebre elevada con escalofríos, acompañado de coluria, acolia e íctero verdínico, hepatomegalia a predominio del lóbulo izquierdo, leucocitosis, elevación importante de la fosfatasa alcalina (FAL), la gammaglutamiltransferasa (GGT) y la bilirrubina.
Se sospechó de diagnósticos como íctero obstructivo por litiasis coledociana, neoplasia de cabeza de páncreas, pero al final se concluyó como posible hepatitis colangiolítica por fármacos.
A los 4 meses presentó cuadro clínico similar, esta vez acompañado de diarreas y dolores osteomioarticulares. Este cuadro se repitió 2 meses después acompañado de insuficiencia suprarrenal aguda caracterizada por hipotensión severa y shock hipovolémico. En todas estas crisis, el paciente fue tratado con antibióticos del tipo de las cefalosporinas de tercera generación, aminoglucósidos y quinolonas.
Las crisis reaparecieron y fue admitido en nuestro centro con fiebre de 39° C, escalofríos, malestar general, dolor en epigastrio e hipocondrio derecho y coluria. Posteriormente, apareció íctero que se fue incrementando. Al realizarle examen físico se le detectó además hepatomegalia dolorosa que rebasaba el reborde costal derecho en 3 cm, taquicardia e hipotensión arterial.
INVESTIGACIONES REALIZADAS
Hemoglobina: 135 g/L, hematócrito: 0,44; leucocitos: 15,4 x 109/L, polimorfonucleares: 87 %; linfocitos:10 %; eosinófilos: 3 %; plaquetas: 168 x 109/L ,VSG: 46 mm/h, ASAT: 342 U/L, ALAT: 648 U/L, GGT: 778 U/L, FAL: 1503 U/L, bilirrubina total: 45,3 mmol/L (BD: 30 mmol/L) , coagulograma: (TP: 17'', TPT con Kaolín: 40''); glucemia: 4,8 mmol/L; creatinina: 88 mmol/L; colesterol: 7,0 mmol/L; triglicéridos: 1,2 mmol/L; proteínas totales: 57g/L; albúmina: 27,7 g/L; antígeno carcinoembrionario-0,285 ng/mL, amilasa sérica-12 U/L, conteo de Addis de 2 h (leucocitos: 13 333 x 10³; hematíes: 26 667 x 10³), 3 hemocultivos y 2 urocultivos, negativos. Marcadores inmunológicos: anticuerpos antimúsculo liso: positivo, anticuerpo antinuclear-positivo patrón difuso, intensidad: 2/4; anticuerpos antimitocondriales: negativo, anticuerpo para hepatitis C: negativo; factor reumatoideo: negativo. Estudios imagenológicos: rayos x de tórax: normal.
En el ultrasonido del abdomen se observó ligera hepatomegalia difusa y marcado engrosamiento de la pared interna del conducto hepático común que dilata ligeramente las vías biliares intrahepáticas, por lo que se sospechó la presencia de una tumoración versus una CEP. En la colangiografía retrógrada endoscópica (CPRE) se demostró la presencia de irregularidad del calibre de las vías biliares, con estenosis y dilataciones que se suceden alternativamente, que hicieron plantear el diagnóstico de la colangitis esclerosante primaria. Se realizó laparoscopia diagnóstica donde se observó que el hígado, la vesícula y el bazo tenían aspecto normal y en la biopsia de hígado se demostró la presencia de depósitos de pigmento biliar dentro de los hepatocitos, con edema e infiltración portal. Se observó además una marcada proliferación fibroblástica pericanalicular e intensa fibrosis concéntrica alrededor de los conductos biliares, no se identificó proliferación neoplásica. El análisis y la revisión histológica evaluaron el estadio de la enfermedad según los criterios de Ludwing y otros18 (figs. 1, 2 y 3).
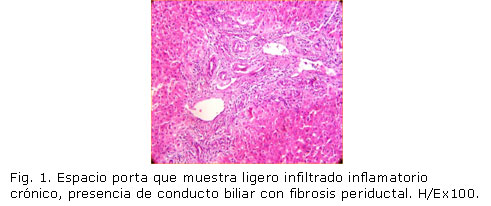

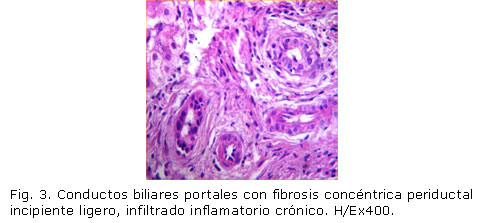
Estadios de la colangitis esclerosante primaria
| Estadio I | Estadio portal. |
| Estadio II | Estadio periportal. Fibrosis periportal o espanciones fibrosas portales. |
| Estadio III | Estadio septal. Fibrosis septal. Puentes de necrosis. |
| Estadio IV | Estadio cirrótico. |
La biopsia hepática que determinó el diagnóstico mostró estadio II de la enfermedad. El paciente realizó tratamiento con ceftriaxona 1 g ev. cada 12 h combinado con gentamicina a dosis de 3 mg/kg/d con buena evolución clínica y analítica.
DISCUSIÓN
Sin lugar a duda, este paciente presenta manifestaciones clínicas de 2 entidades nosológicas diferentes: una colangitis esclerosante primaria, entidad poco frecuente, y un panhipopituitarismo autoinmune. Dos enfermedades que pueden aparecer en el mismo paciente por azar o con una frecuencia mayor, sobre todo si ambas tienen un origen inmunológico.
La CEP afecta sobre todo a adultos y es ligeramente más frecuente en varones que en mujeres, con una relación 2:1. Esta enfermedad se caracteriza por un proceso fibroinflamatorio del árbol biliar que puede afectar los conductos intrahepáticos y extrahepáticos, así como la ampolla de Vater. Se divide en primario, de patogenia autoinmune, y secundario. La forma primaria suele asociarse a enfermedad inflamatoria intestinal (EII) y a otras enfermedades autoinmunes, su curso es progresivo, no existe una terapia efectiva y conduce a colestasis crónica, cirrosis biliar, hipertensión portal e insuficiencia hepática19 y además puede dar lugar al desarrollo de colangiocarcinoma.6,20
Los pacientes con colitis ulcerosa (CUI) y colangitis esclerosante primaria tienen mayor riesgo de displasia y cáncer colorrectal que los que padecen de colitis ulcerosa solamente; el comportamiento clínico de la CUI es más severo cuando se asocia a CEP.
Las enfermedades autoinmunes que más frecuentemente coexisten con la CEP son la diabetes mellitus, la enfermedad celiaca y la enfermedad de Graves.21 También ha sido reportada la asociación con la artritis reumatoidea y su presencia se comporta como un marcador de alto riesgo para la rápida progresión hacia la cirrosis hepática.22 Estos hechos refuerzan su posible naturaleza inmunológica, lo que probablemente esté en relación con un fenómeno de autoinmunidad que requiere la presencia de un terreno genético apropiado, ya que se ha demostrado que esta enfermedad se asocia a ciertos antígenos de histocompatibilidad HLA-A1, B8, DR3, DR4 y DRW52A23 y a la presencia de anticuerpos anticitoplasma de los neutrófilos (ANCA) en el plasma, aunque se postula que estos anticuerpos no intervienen en la patogenia de la CEP y probablemente representan un epifenómeno.24,25 Se observa hipergammaglobulinemia en un tercio de los pacientes y los niveles de IgM se incrementan en 50 % en los casos avanzados.25-27 Otros cambios inmunológicos incluyen disminución de la células T circulantes e incremento de la relación celular CD4/CD8.26 En un número escaso de pacientes se asocia a tiroiditis o fibrosis retroperitoneal. Sin embargo, no hemos encontrado en la literatura revisada asociación con el panhipopitiutarismo autoinmune.
La presentación clínica en este paciente se caracterizó por la progresión rápida de la enfermedad con crisis frecuentes de fiebre elevada con escalofríos, marcada toma del estado general, íctero de instalación rápida y progresivo, leucocitosis con desviación a la izquierda, elevación de la FAL, GGT y bilirrubina y moderada de las transaminasas. Los episodios remitieron rápidamente después de impuesto tratamiento enérgico con antibióticos parenterales, lo que hace sospechar la presencia de una infección bacteriana sobreañadida de las vías biliares (colangitis recidivante bacteriana) complicación temida en esta afección. La identificación de autoanticuerpos (ANCA) apoya el origen inmunológico de esta entidad. Otra característica de este paciente es que en la biopsia de hígado se demostró la existencia de infiltrado linfocitario alrededor de los conductos biliares (pericolangitis). Los pacientes con este tipo de lesión, que afecta los pequeños conductos biliares intrahepáticos, son los que peor pronóstico comportan.17,28
Una vez establecido el diagnóstico se inició tratamiento con ácido ursodesoxicólico que tiende a mejorar la función de las enzimas hepáticas y aunque su efecto sobre las alteraciones histológicas es aun controvertido, existen evidencias que a altas dosis este medicamento es más efectivo y retardan la aparición de los hallazgos colangiográficos, así como la progresión hacia la fibrosis hepática.29,30
Al paciente le fue propuesto el trasplante hepático como única alternativa terapéutica definitiva para su enfermedad, pues la alta recurrencia de las crisis, la progresión hacia la cirrosis hepática y la probabilidad del desarrollo de un colangiocarcinoma le confieren mal pronóstico.31,32
REFERENCIAS BIBLIOGRÁFICAS
1. Angulo P, Lindor KD. Primary sclerosing cholangitis. Hepatology. 1999; 30:325-32.
2. La Russo NF, Wiesner RH, Ludwig J. Colangitis esclerosante primaria. En: Tratado de Hepatología Clínica. T. II. Barcelona: Ed. Masson-Salvat; 1993. p. 893-902. (Ediciones Científicas y Técnicas).
3. Martins EB. Sclerosing cholangitis. Curr Opinión Gastroenterol. 2000;16:444-9.
4. Bambha K, Kim WR, Talwalkar J, Torgerson H, Benson JT, Therneau TM et al. Incidence, clinical spectrum, and outcomes of primary sclerosing cholangitis in a United States community. Gastroenterol. 2003;125:1364-9.
5. Boberg KM, Bergquist A, Mitchell S, Pares A, Rosina F, Broome U, et al. Cholangiocarcinoma in primary sclerosing cholangitis: risk factors and clinical presentation. Scand J Gastroenterol. 2002;37:1205-11.
6. Nashan B, Schlitt HJ. Biliary malignancies in primary sclerosing cholangitis: timing for liver transplantation.Hepathology. 1996 May;23(5);1105-11.
7. Rolf Olsson, Åke Danielsson, Gunnar Järnerot, Eva Lindstrom, Lars Lööf, Peter Rolny et al. Prevalence of primary sclerosing cholangitis in patients with ulcerative colitis. Gastroenterol. 1991;100:1319-23.
8. Shepherd HA, Selby WS, Chapman RW, Nolan D, Barbatis C, McGee JO et al. Ulcerative colitis and persistent liver dysfunction. Q J Med. 1983;52:503-13.
9. Schrumpf E, Fausa O, Elgjo K, Kolmannskog F. Hepatobiliary complications of inflammatory bowel disease. Semin Liver Dis. 1988;8:201-19.
10. Cadahia V, Rodrigo L, Fuentes D, Riestra S de F, Fernandez M. Celiac disease, ulcerative colitis, and primary sclerosing cholangitis in one patient a family study. Rev Esp Enferm Dig. 2005 Dec;97(12):907-13.
11. Barreda F, Contardo C, León A, Navarrete J, Figueroa R, Attanasio F. Colangitis esclerosante primaria asociada a sindrome de Sjogren, fibrosis retroperitoneal y pancreatitis cronica. Reporte de un caso. Rev Gastroenterol Perú. 1989;9(2):106-14.
12. Gow PJ, Fleming KA, Chapman RW. Primary sclerosing cholangitis associated with rheumatoid arthritis and HLA DR4: is the association a marker of patients with progressive liver disease? J Hepatol. 2001;34:631-5.
13. Boberg KM, Aadland E, Jahnsen J, Raknerud N, Stiris M, Bell H. Incidence and prevalence of primary biliary cirrhosis, primary sclerosing cholangitis, and autoimmune hepatitis in a Norwegian population. Scand J Gastroenterol. 1998;33:99-103.
14. Valera JM, Smok G, Fernández M, Brahm J. Evolución de hepatitis autoinmune a colangitis esclerosante primaria. Gastroent Latinoam 2002;13:45-50.
15. Harrison PM. Diagnosis of primary sclerosing cholangitis. J Hepatobiliary Pancreat Surg. 1999,6(4):356-60.
16. Fulcher AS, Turner MA, Franklin KJ, Shiffman ML, Sterling RK, Luketic VA et al. Primary sclerosing cholangitis: evaluation with MR cholangiography: a-case-control study. Radiology. 2000;215:71-80.
17. Olsson R, Danielsson A, Jarnerot G, Lindstrom E, Loof L, Rolny P et al. Primary sclerosing cholangitis: natural history, prognostic factors and survival analysis. Hepatology. 1989;10:430-6.
18. Ludwig J. Surgical pathology of the syndrome of primary sclerosing cholangitis. Am J Surg Pathol. 1989;13:43-9.
19. Farrant JM, Hayllar KM, Wilkinson ML, Karani J, Portmann BC, Westaby D et al. Natural history and prognostic variables in primary sclerosing cholangitis. Gastroenterology. 1991;100:1710-17.
20. Bjornsson E, Kilander A, Olsson RCA. 19-9 and CEA are unreliable markers for cholangiocarcinoma in patients with primary sclerosing cholangitis. Liver. 1999;19:501-8.
21. Saarinen S, Olerup O, Broome U. Increased frequency of autoimmune diseases in patients with primary sclerosing cholangitis. Am J Gastroenterol. 2000;95:3195-9.
22. Gow PJ, Fleming KA, Chapman RW. Primary sclerosing cholangitis associated with rheumatoid arthritis and HLA DR4: is the association a marker of patients with progressive liver disease? J Hepatol. 2001;34:631-5.
23. Donaldson PT. Genetics of liver disease: immunogenetics and disease pathogenesis. Gut. 2004;53:599-608.
24. Terjung B, Worman HJ. Anti-neutrophil antibodies in primary sclerosing cholangitis. Best Pract Res Clin Gastroenterol. 2001;15:629-42.
25. Duerr RH, Targan SR, Landers CJ, Sutherland LR, Shanahan F. Anti-neutrophil cytoplasmic antibodies in ulcerative colitis. Comparison with other colitides/diarrheal illnesses. Gastroenterology. 1991;100:1590-6.
26. Grant AJ, Lalor PF, Salmi M, Jalkanen S, Adams DH. Homing of mucosal lymphocytes to the liver in the pathogenesis of hepatic complications of inflammatory bowel disease. Lancet. 2002;359:150-7.
27. Rodríguez C, Carrión F, Marinovic MA, Chávez E, Preisler J, Pooley F et al. Sindrome de hiper-IgM asociado a colanguitis esclerosante y neoplasia vesicular. Caso clinico. Rev Med Chil. 2003 Mar;131(3):303-8.
28. Broome U, Olsson R, Loof L, Bodemar G, Hultcrantz R, Danielsson A et al. Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut. 1996;38:610-15.
29. Harnois DM, Angulo P, Jorgensen RA, Larusso NF, Lindor KD. High-dose ursodeoxycholic acid as a therapy for patients with primary sclerosing cholangitis. Am J Gastroenterology. 2001;96:1558-62.
30. Olsson R, Boberg KM, de Muckadell OS, Lindgren S, Hultcrantz R, Folvik G et al. High-dose ursodeoxycholic acid in primary sclerosing cholangitis: a 5-year multicenter, randomized, controlled study. Gastroenterology. 2005;129:1464-72.
31. Bjoro K, Schrumpf E. Liver transplantation for primary sclerosing cholangitis. J Hepatol. 2004;40:570-2.
32. Graziadei IW, Wiesner RH, Marotta PJ, Porayko MK, Hay E, Charlton MR et al. Long-term results of patients undergoing liver transplantation for primary sclerosing cholangitis. Hepatology. 1999;30:1121-7.
Recibido: 9 de octubre de 2007.
Aprobado: 3 de enero de 2008.
Dr. Rolando Rodríguez Fernández. Hospital Clinicoquirúrgico "Hermanos Ameijeiras", San Lázaro No. 701 entre Belascoaín y Marqués González, Centro Habana, Ciudad de La Habana, Cuba. Habana 3, CP 10300.

