










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión impresa ISSN 0034-7531
Rev Cubana Pediatr vol.86 no.1 Ciudad de la Habana abr.-jun. 2014
Revista Cubana de Pediatría. 2014; 86(1)
ARTÍCULO ORIGINAL
Anomalías congénitas asociadas a la atresia esofágica
Esophageal atressia-associated congenital anomalies
MSc. Dr. Roberto Reyes Rodríguez, Dr. José Muñiz Escarpanter, Dr. Ismael Polo Amorín, MSc. Dr. Manuel Alejandro Alvaredo Soria, Dr. Abel Armenteros García, Dra. Neisy María Hernández Fernández
Hospital Pediátrico Universitario "José Luis Miranda". Villa Clara, Cuba.
RESUMEN
Introducción: la atresia esofágica se presenta en 1 de cada 3 000 a 4 500 neonatos vivos. Existe un ligero predominio en los varones, aunque esto no es un hallazgo universal y tal vez no sea cierto para todas las variedades. Con el aumento en la supervivencia de estos niños operados de atresia esofágica, las anomalías asociadas han adquirido mayor significación; más de la mitad de estos tienen una o más anomalías acompañantes.
Objetivo: caracterizar las anomalías asociadas a esta entidad.
Métodos: se realizó un estudio observacional descriptivo transversal de todos los casos diagnosticados de atresia esofágica con o sin fístula traqueoesofágica, en el periodo comprendido desde enero de 2000 hasta diciembre de 2011, en el Hospital Pediátrico Docente Provincial "José Luis Miranda" de Santa Clara, Cuba.
Resultados: predominaron las anomalías congénitas cardiovasculares, seguido de las malformaciones digestivas y respiratorias. La anomalía asociada más frecuente, junto a la comunicación interatrial, fue la malformación anorrectal. Hubo 1 caso de atresia duodenal, así como otro de agenesia diafragmática. No hubo asociaciones, y la única trisomía presentada fue una 21. Los pacientes con anomalías congénitas no cardiovasculares tienen 2,1 veces más probabilidades de morir, que los que no se asocian a este tipo de anomalías. La mortalidad global en esta serie fue del 46,9 %.
Conclusiones: la presencia de una anomalía congénita eleva la mortalidad en estos pacientes, sobre todo, las no cardiovasculares mayores y las cardiovasculares críticas.
Palabras clave: atresia esofágica, recién nacido, anomalías congénitas, mortalidad.
ABSTRACT
Introduction: esophageal atresia occurs in one out of 3 000 or 4 500 livebirths. Males are slightly predominant, but this is not a universal finding and maybe it is not valid for all varieties. With increasing survival rates of children operated on from esophageal atresia, the associated anomalies have become more significant since more than half of these children suffer one or more accompanying anomalies.
Objective: to characterize the esophageal atresia-associated anomalies.
Methods: an observational, cross-sectional and descriptive study of all the cases diagnosed with esophageal atresia, with or without tracheal esophageal fistula, from January 2000 to December 2011 was conducted in "José Luis Miranda" provincial pediatric teaching hospital in Santa Clara, Villa Clara province, Cuba.
Resultados: congenital cardiovascular anomalies were prevailing, followed by digestive and respiratory malformations. The most frequent one, together with interatrial communication, was anorectal malformation. There was a case of duodenal atresia as well as another case of diaphragmatic agenesis. There was no association between them. The patients with non-cardiovascular congenital anomalies are 2.1 times more likely to die than those unrelated to this type of anomaly. The global mortality rate for this series was 46.9 %.
Conclusions: the existence of congenital anomaly raises the mortality likelihood for these patients, mainly in cases of major non-cardiovascular and critical cardiovascular anomalies.
Keywords: esophageal atresia, newborn, congenital anomalies, mortality.
INTRODUCCIÓN
La atresia esofágica (AE) con o sin fístula traqueoesofágica (FTE) se presenta en 1 de cada 3 000 a 4 500 neonatos vivos.1,2 Existe un ligero predominio en los varones, aunque esto no es un hallazgo universal, y tal vez no sea cierto para todas las variedades.1,3 Existen varias clasificaciones que ayudan a determinar el pronóstico de estos niños, entre las que se encuentra la de Waterston,3 la de Montreal2 y la de Spitz.3 La más conocida es la de Waterston, quien establece el pronóstico en función de 3 factores: bajo peso al nacer, presencia de neumonía y malformaciones congénitas asociadas.3
La supervivencia por lo general es buena, en la actualidad es superior a 90 %. La mejoría en la supervivencia no solo se debe al tratamiento quirúrgico, sino a los avances en los cuidados intensivos neonatales, particularmente el apoyo ventilatorio y nutricional que requieren estos pacientes. Los niños con mayor riesgo de muerte son aquellos con peso al nacimiento menor de 1 500 g, con malformaciones cardiacas o anomalías cromosómicas. Las muertes tempranas son resultado de malformaciones cardiacas o cromosómicas, y las tardías, por lo general, son secundarias a complicaciones respiratorias.2
Con el aumento en la supervivencia de estos niños operados de AE, las anomalías asociadas han adquirido mayor significación, ya que más de la mitad de estos tienen una o más anomalías acompañantes, más frecuentes en la AE con FTE, y menos frecuentes en la FTE sin AE (variedad en N).4-9
Otro capítulo de las malformaciones asociadas que cobra gran interés son las malformaciones múltiples. Reciben su nombre con las siglas en idioma inglés de los defectos que las integran, y entre las más conocidas se encuentran el VATER, que incluye defectos vertebrales, la malformación anorrectal, la FTE, la AE y anomalía renal o displasia radial. Si se le agregan un defecto cardiaco y de la pierna, se conoce entonces como VACTERL. Del 5 al 17 % de los pacientes con AE cumplen los criterios para esta asociación.10 Otra de las asociaciones conocidas son el CHARGE, que incluye, coloboma, retardo del crecimiento y desarrollo, hipoplasia genital y anomalías auriculares, así como algunas alteraciones genéticas, como las trisomías 13, 18, 21, y el síndrome de Potter, con agenesia renal e hipoplasia pulmonar.7-11 En nuestro medio se desconoce cuál es el comportamiento de estas anomalías que se asocian a la AE, así como su influencia real en la mortalidad por esta causa. Es por ello que se realiza este estudio, para caracterizar las anomalías asociadas a esta entidad.
MÉTODOS
Se realizó un estudio observacional descriptivo transversal de todos los casos diagnosticados de AE con o sin FTE, en el periodo comprendido desde enero de 2000 hasta diciembre de 2011, en el Hospital Pediátrico Docente Provincial "José Luis Miranda" de Santa Clara, Cuba. El universo de estudio estuvo integrado por el total de nacidos vivos con diagnóstico de AE y/o FTE en ese periodo, y atendidos en este hospital. Se realizó un muestreo intencional no probabilístico, teniendo en cuenta los criterios de inclusión siguientes:
1. Diagnóstico positivo de AE y/o FTE.
2. Tratado quirúrgicamente en nuestro centro.
La muestra coincidió con el universo. Las fuentes de información fueron: las historias clínicas pediátricas, maternas y genéticas, e informes operatorios. Las variables estudiadas fueron: peso al nacer, sexo, edad gestacional, cardiopatía congénita (malformación cardiaca asociada clasificada en críticas, potencialmente críticas y no críticas), anomalías congénitas no cardíacas (malformaciones asociadas, diferentes a las cardíacas, clasificadas en menores mayores) y asociaciones y/o trisomías, así como la mortalidad.
El análisis estadístico se realizó con el programa estadístico SPSS (software SPSS para Windows v15.0). Las variables estudiadas se expresaron como valor absoluto y porcentaje. La asociación de variables cualitativas entre sí se realizó por medio del estadígrafo x2. Los niveles de significación fueron fijados en:
- Muy significativo: p< 0,01.
- Significativo: p< 0,05.
Se determinó, asimismo, el riesgo relativo de cada variable con respecto a la variable muerte. Para valorar el riesgo de mortalidad intrahospitalaria de los factores estudiados, se realizó un análisis de regresión logística binaria, utilizando el método denominado "condicional hacia atrás", con una probabilidad de entrada de 0,05 y una probabilidad de salida de 0,01. La muerte intrahospitalaria fue la variable dicotómica dependiente. Las variables independientes estudiadas fueron las que resultaron significativas en el análisis univariado. La regresión logística valora las variables independientes simultáneamente, seleccionando primero aquella que se relaciona mejor con el pronóstico. En ciclos consecutivos el programa valora el resto de variables, y permite su entrada en la función si aumenta significativamente la precisión del modelo.
RESULTADOS
La media del peso al nacer fue de 2 460 g con pesos que oscilaron entre 1 100 y 4 210 g. La edad gestacional promedio fue de 37 semanas, con valores máximos y mínimos de 42,6 y 29,4 semanas respectivamente. Hubo un predominio de pacientes del sexo femenino (21).
En las tablas 1 y 2, se aprecia un predominio de las anomalías cardiacas con 14 variedades presentes en 8 pacientes, seguidas de las anomalías digestivas y respiratorias. Individualmente la anomalía más frecuente fue la comunicación interatrial (CIA) y la malformación anorrectal en 3 casos cada una. No hubo asociaciones, y la única trisomía observada fue una 21.
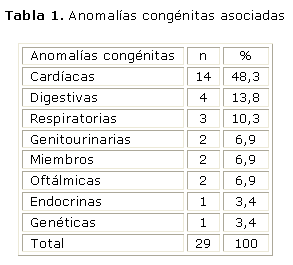
Tabla 2. Tipos de anomalías congénitas asociadas
| Anomalías congénitas | N | % | ||
| Cardíacas | Críticas | Hipoplasia de cavidades izquierdas | 1 | 3,4 |
| Coartación de la aorta (CoAo) severa | 1 | 3,4 | ||
| Atresia tricuspídea | 1 | 3,4 | ||
| Transposición de grandes vasos | 1 | 3,4 | ||
| Inversión ventricular | 1 | 3,4 | ||
| Defecto de septación auriculoventricular completo | 1 | 3,4 | ||
| Doble emergencia del ventrículo derecho | 1 | 3,4 | ||
| | Comunicación interatrial (CIA) | 3 | 10,3 | |
| Persistencia del conducto arterioso | 2 | 6,9 | ||
| Comunicación interventricular (CIV) | 2 | 6,9 | ||
| Subtotal | 14 | 48,3 | ||
| No cardíacas | | Malformación anorrectal | 3 | 10,3 |
| Atresia duodenal | 1 | 3,4 | ||
| Agenesia diafragmática | 1 | 3,4 | ||
| Hipoplasia pulmonar | 1 | 3,4 | ||
| Hipoplasia suprarrenal bilateral severa | 1 | 3,4 | ||
| Atresia coanas | 1 | 3,4 | ||
| Trisomía 21 | 1 | 3,4 | ||
| | Anomalías de los dedos de la mano | 2 | 6,9 | |
| Hipospadias | 1 | 3,4 | ||
| Genitales ambiguos | 1 | 3,4 | ||
| Fisura palpebral | 1 | 3,4 | ||
| Microftalmia | 1 | 3,4 | ||
| Subtotal | 15 | 51,7 | ||
| Total | 29 | 100 | ||
En la tabla 3 se observa que existe una relación significativa entre la mortalidad y las variables siguientes: anomalías congénitas cardiovasculares (CDV), anomalías congénitas CDV críticas, anomalías congénitas no CDV y anomalías congénitas no CDV mayores.
La tabla 4 muestra la variable que resultó significativa para predecir mortalidad después del análisis multivariado, esta fue la presencia de anomalías congénitas no CDV.
DISCUSIÓN
La media del peso al nacer de nuestra serie coincide con otras series publicadas en América Latina,5 Estados Unidos6 y Europa.8 Solo un trabajo publicado en México12 muestra una media de peso de 1 950 g (rango 1 450 y 3 550 g), inferior a la nuestra.
En la mayoría de las series se ha observado, por el contrario, un ligero predominio del sexo masculino.5,6 Otras12 no han encontrado diferencias significativas, y en una serie13 de 20 pacientes pediátricos portadores de AE, 11 (55 %) fueron del sexo femenino y 9 (45 %) del sexo masculino.
La asociación de una o varias malformaciones en la AE es un fenómeno frecuente que alcanza cifras que oscilan entre el 30 y el 70 %.11 La frecuencia de malformaciones congénitas asociadas a AE en este estudio fue de 37,5 % (12 pacientes). Uno reciente, realizado en Holanda, informó una prevalencia de 66 % en los últimos años,12 mientras otro declaró un valor cercano al 50 %.11 Otra serie encontró que la prevalencia de malformaciones congénitas fue del 45 % y que la malformación cardiovascular es la más frecuente (29 %).13 De entre todas las malformaciones, la cardiaca se ha descrito como la más repetida (entre el 13 y el 31 %),11 en cuyo intervalo no está el valor encontrado en este estudio de 48,3 %, sin embargo la proporción de malformación congénita cardiaca (48,3 %), continúa siendo similar al 51 % encontrado en una reciente serie de casos.13 Como cita Spitz en su experiencia,7 "los defectos del septo ventricular son las principales causas de malformación cardiaca en la AE", hecho que se corrobora con los hallazgos de este artículo. El defecto del septo ventricular ocurrió en 22,3 % de los casos en otro reciente estudio.13
En cuanto al resto de las malformaciones, la más común reportada por la literatura14-17 es la malformación anorrectal. La literatura15-17 reporta las asociaciones en porcentajes tan altos como el 18-30 % de los casos; en cambio, en la nuestra no hubo ninguna asociación.
La mortalidad en esta serie fue de 46,9 %, superior a series internacionales que reportan una supervivencia superior al 90 %.11-13 Por lo general, estas son publicaciones de países desarrollados que, lógicamente, tienen mejores resultados, pues cuentan con más experiencia y mayor cantidad de recursos. Por otro lado, la mortalidad durante el período estudiado se comportó de manera similar a otros estudios realizados en nuestro país,16 y a un trabajo publicado por Rokitansky y otros7 en Viena, que incluye 17 años de experiencia con una mortalidad global del 41,3 %.
Todas las anomalías cardiovasculares críticas de este estudio fallecieron, según muestra la tabla 3, no así para las no críticas, en las que el 50 % sobrevivió. Es por eso que al realizar el análisis univariado se encontró que la presencia de una anomalía cardiovascular no crítica no está relacionada con la mortalidad, no así para las anomalías cardiovasculares críticas, que sí resultaron significativas. La explicación a este fenómeno sería la siguiente: la mayoría de las veces la anomalía cardiovascular no es ductus dependiente, como ocurre en las no críticas (defectos septales sin repercusión hemodinámica), y esto permite efectuar la corrección completa de la atresia esofágica. Cuando se detecta una anomalía cardiovascular ductus dependiente (es el caso de las críticas), el manejo depende del estado general del paciente, ya que se modifica la hemodinamia del neonato, y por tanto, aumentan las probabilidades de fallecer. En la serie estudiada el riesgo es de 5,8 veces.
El grupo de Montreal,14,15 en 1996, describe el valor de las anomalías congénitas no cardiovasculares como factor pronóstico de mortalidad. En este estudio la mayoría de los pacientes presentó una anomalía congénita mayor 7 de 8 recién nacidos, según presenta la tabla 3 y de estos, 6 fallecieron. El análisis univariado demostró que los pacientes con una anomalía congénita no cardiovascular, se relacionan significativamente con la mortalidad, y tienen 10,6 veces más riesgo de morir.
Los pacientes con una anomalía cardiovascular mayor tienen, por definición, un riesgo elevado de fallecer, o requieren corregir el defecto antes o simultáneamente con la corrección de la atresia esofágica, lo cual, como es de suponer, aumenta las probabilidades de morir del recién nacido, ya que no solo prolonga el tiempo anestésico, sino que también prolonga la recuperación posoperatoria de estos pacientes con la aparición de complicaciones relacionadas con la anomalía asociada además. En la tabla 4 se demuestra cómo estos pacientes tienen 2,1 veces más probabilidades de morir, que los que no se asocian a este tipo de anomalías.
Se puede concluir planteando que predominan las anomalías congénitas CDV, y que son las más frecuentes los defectos septales y la persistencia del conducto arterioso, seguido de las malformaciones digestivas y respiratorias. La anomalía asociada más frecuente, junto a la CIA, es la malformación anorrectal. Los pacientes con anomalías congénitas no CDV tienen 2,1 veces más probabilidades de morir que los que no se asocian a este tipo de anomalías.
REFERENCIAS BIBLIOGRÁFICAS
1. Robb A, Lander A. Oesophageal atresia and tracheooesophageal fistula. Surgery (Oxford). 2008 jul;25(7):283-6.
2. Holder TM, Clout DT, Lewis Jr JE, Pilling 4th GP. Esophageal atresia and tracheoesophageal fistula: a survey of its members by the surgical section of the American Academy of Pediatrics. Pediatrics. 2008;34:542.
3. De Jong EM, De Haan M, Gischler SJ, Hop W, Cohen- Overbeek TE, Bax N, et al. Pre and postnatal diagnosis and outcome of fetuses and neonates with esophageal atresia and tracheoesophageal fistula. Prenat Diagn [serie en Internet]. 2010 [citado 10 de marzo de 2012];30(3). Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/20112230
4. Nasr A, McNamara PJ, Mertens L, Levin D, James A, Holtby H, et al. Is routine preoperative 2-dimensional echocardiography necessary for infants with esophageal atresia, omphalocele, or anorectal malformations? J Pediatr Surg [serie en Internet]. 2010 [citado 5 de enero de 2011];45(5). Disponible en: http://www.jpedsurg.org/article/abstracts?terms1=&terms2=&terms3
5. Villegas-Álvarez F, González-Zamora JF, Braun-Roth G, López-Corella E. Causas de muerte de un grupo de niños con atresia de esófago sometidos a autopsia. Perinatol Reprod Hum [serie en Internet]. 2008 [citado 10 de marzo de 2012];17(1). Disponible en: http://new.medigraphic.com/cgi-bin/resumenMain.cgi?IDARTICULO=21231&IDPUBLICACION=2156&IDREVISTA=76
6. Delius RE, Wheatley MJ, Coran AG. Etiology and management of respiratory complications alter repair of esophageal atresia with tracheoesophageal fistula. Surgery. 2010;112:527-32.
7. Rokitansky A. Recent evaluation of prognostic risk factors in esophageal atresia. A multicenter review of 223 cases. J Ped Surg. 2008;3:196-201.
8. Agarwala S, Bhatnagar V, Bajpai M, Gupta DK, Mitra DK. Factors contributing to poor results of treatment of esophageal atresia in developing countries. Pediatr Surg Int. 2009;11:312-15.
9. Saing H, Mya GH, Cheng W. The involvement of two or more systems and the severity of associated anomalies significantly influence mortality in esophageal atresia. J Pediatr Surg. 2008;33:1596-8.
10. Choudhury SR, Ashcraft KW, Sharp RJ. Survival of patients with esophageal atresia: influence of birth weight, cardiac anomaly and late respiratory complications. J Pediatr Surg. 2009;34:70-4.
11. Gupta DK, Arora M, Srinivas M. Azygos vein anomaly: the best predictor of a long gap in esophageal atresia and tracheoesophageal fistula. Pediatr Surg Int. 2011;17:101-3.
12. Tabaré Martínez-Carreño U, Paniagua-Morgan F, Victoria-Morales G, de la Torre-Mondragon L. Complicaciones postoperatorias en los pacientes con atresia esofágica tipo III. Revista Mexicana de Cirugía Pediátrica [serie en Internet]. 2008 [citado 10 de marzo de 2012];15(3).
Disponible en: http://new.medigraphic.com/cgi-bin/resumen.cgi?IDREVISTA=91&IDARTICULO=26716&IDPUBLICACION=2738
13. Schulz AC, Bartels E, Stressig R, Ritgen J, Schmiedeke E, Mattheisen M, et al. Nine new twin pairs with esophageal atresia: A review of the literature and performance of a twin study of the disorder. Biths Defects Res A Clin Mol Teratol [serie en Internet]. 2012 [citado 15 de mayo de 2012];94(3). Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/22287212?dopt=Abstract
14. Jakubsons L, Paz F, Aleja Paul R. Harris D, Bertrand P. Atresia esofágica y fístula traqueoesofágica. Evolución y complicaciones postquirúrgicas. Rev Chil Pediatr. 2010;81(4):339-46.
15. Fierro C, Caro M, Anzieta J, Butte JM, González P, Apablaza JP. Atresia esofágica. Manejo quirúrgico en el Hospital Clínico Regional de Valdivia. Cuad Cir. 2008;16:20-5.
16. Xiaomei L, Aras-Lopez R, Martinez L, Tovar JA. Lung hypoplasia in rats with esophageal atresia and tracheoesophageal fistula. Pediatr Res. 2012;71(3):235-40.
17. Castro Guevara JE, Hernández Moore E, Bueno Rodríguez JC, Aguilar Atanay D. Comportamiento de la atresia esofágica en Camagüey. Archivo Médico de Camagüey [serie en Internet]. 2005 [citado 10 de marzo de 2012];9(4). Disponible en: http://www.amc.sld.cu/amc/2005/v9n4/1003.htm
Recibido: 2 de febrero de 2013.
Aprobado: 1º de agosto de 2013.
Roberto Reyes Rodríguez. Hospital Pediátrico Universitario "José Luis Miranda". Rodrigo # 453, entre Roble y Salustiano Pedraza, Reparto Raúl Sancho, municipio Santa Clara. Villa Clara, Cuba. Correo electrónico: marisleinysad@edu.vcl.sld.cu

{kind=link}
{kind=link}