Presentación de caso
Síndrome de Freeman-Sheldon
0000-0002-4628-7698Liset Betancourt Castellanos
1
*
, 0000-0002-0637-3217Idarmis Jiménez Torres
2
, 0000-0001-7911-8906Marioneya Izaguirre Bordelois
1
, 0000-0002-7019-7182Denny Betancourt Castellanos
3
1Universidad Técnica de Manabí. Manabí, Ecuador.
2Hospital Gineco Obstétrico “Isidro Ayora”. Pichincha, Ecuador.
3Hospital Provincial “Antonio Luaces Iraola”. Ciego de Ávila, Cuba.
RESUMEN
Introducción:
El síndrome de Freeman-Sheldon es un síndrome hereditario raro, de severidad variable que afecta principalmente la cara, manos y pies, sin preferencia de género, étnica o geográfica.
Objetivo:
Caracterizar clínicamente a un paciente con síndrome Freeman-Sheldon.
Presentación del caso:
Niña ecuatoriana de 6 años de edad, hija de madre de 43 años y padre de 42 años, la cuarta de 6 hermanos, todos sanos, no historia de consanguinidad. La cual presenta cara parecida a una máscara, ojos hundidos, puente nasal ancho, boca pequeña con apariencia de silbador, hoyuelo cutáneo en mentón en forma de H, defecto en las manos, contractura de los dedos con desviación cubital y pies equinovaro, dificultad para la marcha y baja talla.
Conclusiones:
El síndrome de Freeman-Sheldon es un síndrome raro que afecta principalmente la cara y las extremidades de los pacientes, cuyo diagnóstico clínico es posible luego de un examen físico exhaustivo.
Palabras-clave: síndrome de Freeman-Sheldon; síndrome de Freeman-Burian; displasia craneocarpotarsal; distrofia craneocarpotarsal; síndrome craneofacial; artrogriposis distal tipo 2A; síndrome de cara de silbido
ABSTRACT
Introduction:
Freeman-Sheldon syndrome is a rare hereditary syndrome of varying severity that mainly affects the face, hands and feet, without gender, ethnic or geographical preference.
Objective:
Clinically characterize a patient with Freeman-Sheldon syndrome.
Presentation of the case:
Ecuadorian girl, 6 years old, daughter of mother of 43 years and father of 42 years, the fourth of 6 brothers, all healthy, not history of consanguinity. She presents mask-like face, sunken eyes, wide nasal bridge, small mouth with the appearance of a whistler, skin dimple on the chin in the shape of an H, defect in the hands, contracture of the fingers with ulnar deviation and clubfoot, also walking difficulty and short height.
Conclusions:
Freeman-Sheldon syndrome is a rare syndrome that mainly affects the face and limbs of patients, whose clinical diagnosis is possible after a thorough physical examination.
Key words: Freeman-Sheldon syndrome; Freeman-Burian syndrome; craniocarpotarsal dysplasia; craniocarpotarsal dystrophy; craniofacial syndrome; distal arthrogryposis type 2A; whistling face syndrome
Introducción
El síndrome de Freeman-Sheldon (FSS) (OMIM 193700), es un síndrome craneofacialmiopático congénito raro, de carácter hereditario, con severidad variable.1,2) Afecta principalmente la cara, manos y pies3) e incluye dentro de sus manifestaciones, anomalías óseas, articulaciones contracturadas, y rasgos faciales típicos. Descrito por primera vez en 1938 por Freeman y Sheldon, en su comunicación de dos casos clínicos a los que definieron como “distrofia cráneo-carpo-tarsal”,4) sin embargo, las características alteraciones faciales condicionaron que Burian en 1963 acuñara el término de “síndrome en cara de silbido”.5) Se ha logrado determinar que puede llegar a ser causado por una mutación en el gen MYH3.6,7
La frecuencia sigue siendo desconocida, debido a la incertidumbre diagnóstica,8) sin embargo, algunos autores plantean una prevalencia de 0,9 por 1 millón y no parece haber preferencia de género, étnica o geográfica.1) Se han publicado menos de 100 casos en la literatura.3) El objetivo de este trabajo es caracterizar clínicamente a un paciente con síndrome Freeman-Sheldon.
Presentación del caso
Niña ecuatoriana, de 6 años de edad, que asiste en compañía de su madre a la consulta de genética clínica por primera vez, por presentar rasgos dismórficos faciales y discapacidad fisicomotora. Hija de madre de 43 años y padre de 42 años, la cuarta de 6 hermanos, todos sanos. No historia de consanguinidad.
Examen físico. Cara: frente alta y cara parecida a una máscara, ojos hundidos, puente nasal ancho, telecanto, epicanto, estrabismo convergente y blefarofímosis, boca pequeña con apariencia de silbador, hoyuelo cutáneo en mentón en forma de H y apiñamiento dental (Fig. 1).
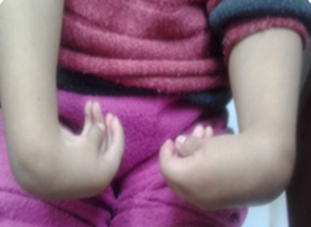
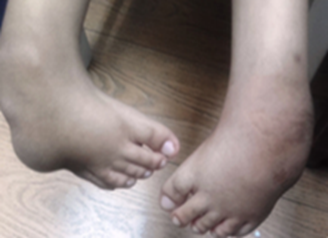
Sistema osteomioarticular: defectos en las manos, contractura de los dedos con desviación cubital de muñecas y dedos (Fig. 2), y pies equinovaro (Fig. 3). Hipotrofia muscular en extremidades superiores e inferiores, contracturas de hombros, codos, cadera y rodillas, dificultad para la marcha y baja talla.
En la actualidad, la paciente está pendiente para la realización de una cirugía correctiva en miembros inferiores y presenta trastornos severos del lenguaje y dificultad para la alimentación debido a la intervención tardía. Se obtuvo el consentimiento del familiar para la toma y publicación de las imágenes.
Exámenes complementarios: cariotipo: 46, XX (20 metafases).
Discusión
La paciente fue diagnosticada con el FSS luego de un examen físico exhaustivo que destaca un estado dismórfico característico, con anomalías óseas y contracturas articulares con facie típica de silbido; correspondiéndose con lo expuesto en la literatura acerca de que estos pacientes presentan microstomía, apariencia de silbato (labios fruncidos), defecto del mentón en forma de H o V y prominentes pliegues nasolabiales, malformaciones en las extremidades con desviación cubital de la mano y talipes equinovarus.1,2,9,10) Otros autores11 plantean la existencia de apiñamiento dental y pérdida de la audición, sin embargo, nuestra paciente no presenta trastornos auditivos.
Se plantea además, que la inteligencia es normal8 en pacientes con este síndrome, lo cual no se ha definido en nuestro caso, pero si hemos podido constatar que comprende las preguntas que se le realizan.
Se ha observado consanguinidad en familia con casos de este síndrome, por lo que se sospecha la presencia de un patrón de herencia del tipo autosómico recesivo;5,12) sin embargo, la mayoría de los casos de FSS son esporádicos; el patrón de herencia más descrito es el autosómico dominante y se cree que son mutaciones en el gen de la cadena pesada de la miosina embrionaria (MYH3) en 17p13.1-pter.3,12,13
En nuestra paciente no hay historia de consanguinidad, ni historia familiar de defectos congénitos similares, lo que sugiere que se trata de un caso esporádico debido a una mutación de novo.
Casi todas las mutaciones de MYH3 interfieren con la actividad catalítica de la miosina, quien está involucrada en el movimiento celular y junto a la actina, son los principales componentes de las fibras musculares y son importantes para la contracción muscular.6 Existe correlación genotipo y fenotipo en la mutación p.T178I, la cual ocasiona graves contracturas faciales y escoliosis congénitas.14) En nuestra paciente está pendiente la realización del estudio molecular.
Es importante el diagnóstico precoz del FSS y su seguimiento multidisciplinario, para evitar posibles complicaciones y así poder mejorar la calidad de vida del paciente.15) En nuestro caso, la intervención ha sido tardía por lo que la paciente presenta trastornos severos del lenguaje y dificultad para la alimentación.
La hipertermia maligna e intubaciones difíciles, son complicaciones que se pueden presentar durante las intervenciones quirúrgicas en estos pacientes,8aspecto que se está tomando en cuenta en la planificación de la cirugía correctiva en miembros inferiores a la que debe ser sometida nuestra paciente.
Concluimos que el síndrome de Freeman-Sheldon es un síndrome raro que afecta principalmente la cara y las extremidades de los pacientes, cuyo diagnóstico clínico es posible luego de un examen físico exhaustivo.
Referencias bibliográficas
1.
Poling MI, Dufresne CR, Chamberlain RL. Freeman-Burian syndrome. Orphanet J Rare Dis. 2019 [acceso 16/07/2019];14(1):14. Disponible en: Disponible en: https://translate.googleusercontent.com/translate_c?depth=1&hl=es&prev=search&rurl=translate.google.com&sl=en&sp=nmt4&u=https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6327538/&xid=25657,15700023,15700186,15700190,15700256,15700259,15700262&usg=ALkJrhikZWAx2GM2lEKClvHE3N3Ig_7b0g
1.
[ Links ]
2.
Aguilar HA, Bustos G, Vargas J, González A, Camacho JF, Barajas-Gamboa JS. Impacto de enfermedades raras en la práctica médica. Síndrome de Freeman-Sheldon. Rev ColombCir Plástica Reconstr. 2015 [acceso 17/07/2019];21(1). Disponible en: Disponible en: https://www.ciplastica.com/sccp07-julio-2015
2.
[ Links ]
3.
Gurjar VV, Parushetti A, Gurjar M. Freeman-Sheldon Syndrome Presenting with Microstomia: A Case Report and Literature Review. J Maxillofac Oral Surg. 2013 [acceso 25/09/2019];12(4):395-9. Disponible en: Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3847022/
3.
[ Links ]
4.
Freeman EA, Sheldon JH. Cranio-carpo-tarsal dystrophy. Arch Dis Child. 1938 [acceso 14/08/2021];13(75):277-83. Disponible en: Disponible en: https://adc.bmj.com/content/13/75/277
4.
[ Links ]
5.
Alonso JL, Ali K. Síndrome de Freeman-Sheldon: manifestaciones clínicas y manejo anestésico y quirúrgico. An Esp Pediatr. 2002 [acceso 16/07/2019];56(2):175-9. Disponible en: Disponible en: https://www.analesdepediatria.org/es-sindrome-freeman-sheldon-manifestaciones-clinicas-manejo-articulo-resumen-S1695403302789510
5.
[ Links ]
6.
Toydemir RM, Rutherford A, Whitby FG, Jorde LB, Carey JC, Bamshad MJ. Mutations in embryonic myosin heavy chain (MYH3) cause Freeman-Sheldon syndrome and Sheldon-Hall syndrome. Nat Genet. 2006 [acceso 16/07/2019];38(5):561-5. Disponible en: Disponible en: https://www.nature.com/articles/ng1775?cacheBust=1509107145158
6.
[ Links ]
7.
Racca AW, Beck AE, McMillin MJ, Korte FS, Bamshad MJ MJ, Regnier M. The embryonic myosin R672C mutation that underlies Freeman-Sheldon syndrome impairs cross-bridge detachment and cycling in adult skeletal muscle. Hum Mol Genet. 2015 [acceso 25/09/2019];24(12):3348-58. Disponible en: Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/25740846
7.
[ Links ]
8.
McCormick RJ, Poling MI MI, Chamberlain RL. Bilateral patellar tendon-bearing Symes-type prostheses in a severe case of Freeman-Sheldon syndrome in a 21-year-old woman presenting with uncorrectable equinovarus. BMJ Case Rep. 2015 [acceso 25/09/2019];2015(bcr2015211338). Disponible en: Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4513510/
8.
[ Links ]
9.
Onal O, Gumus I, Sari M, Zora ME, Acar MA. The Obv-Eas Method: An Easy Way to Facilitate Fiberoptic Intubation in Pediatric Patients: Case of an Infant with Freeman-Sheldon Syndrome. Anesthesiol Pain Med. 2018;8(5) [acceso 16/07/2019]:e81451. Disponible en: Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6240828/
9.
[ Links ]
10.
Kamal G, Shah SB, Gupta A. Anesthesia Challenges in the Management of Freeman-Sheldon Syndrome: Report of Two Cases and Literature Review. AANA J. 2020 [acceso 24/09/2019];88(1):35-8. Disponible en: Disponible en: https://pubmed.ncbi.nlm.nih.gov/32008616/
10.
[ Links ]
11.
Ali AM, Mbwasi RM, Kinabo G, Kamsteeg EJ, Hamel BC, Dekker MCJ. Freeman-Sheldon Syndrome: First Molecularly Confirmed Case from Sub-Saharan Africa. Case Rep Genet. 2017 [acceso 25/09/2019];5. Disponible en: Disponible en: https://www.hindawi.com/journals/crig/2017/9327169/cta/
11.
[ Links ]
12.
Rashid AKMM, Ahmmad F, Rupa HS. Freeman-Sheldon Syndrome - A Case of Rare Observation. J Ped Moth Care. 2016 [acceso 16/07/2019];2(1):106. Disponible en: Disponible en: https://www.elynspublishing.com/journal/article/freeman-sheldon-syndrome-a-case-of-rare-observation
12.
[ Links ]
13.
Das S, Kumar P, Verma A, Maiti TK, Mathew SJ. Myosin heavy chain mutations that cause Freeman-Sheldon syndrome lead to muscle structural and functional defects in Drosophila. Dev Biol. 2019 [acceso 13/08/2021];449(2):90-8. Disponible en: Disponible en: https://pubmed.ncbi.nlm.nih.gov/30826400/
13.
[ Links ]
14.
Reina-Ramírez F, Jiménez-Cardozo N, Hurtado-Villa P. Síndrome de Freeman Sheldon, reporte de caso y revisión de la literatura. Salutem Scientia Spiritus. 2017 [acceso 14/08/2021];3(1):46-51. Disponible en: Disponible en: https://revistas.javerianacali.edu.co/index.php/salutemscientiaspiritus/article/view/1726
14.
[ Links ]
15.
Santana Hernández EE. Síndrome Freeman- Sheldon. Presentación de un caso. Medimay. 2021 [acceso 14/08/2021];28(1):123-8. Disponible en: Disponible en: http://revcmhabana.sld.cu/index.php/rcmh/article/view/1949
15.
[ Links ]











 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink