










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Obstetricia y Ginecología
versión impresa ISSN 0138-600X
Rev Cubana Obstet Ginecol vol.39 no.3 Ciudad de la Habana jul.-sep. 2013
REVISIÓN BIBLIOGRÁFICA
Lipoxinas inducidas por la aspirina: una alternativa para modular los procesos proinflamatorios en la preeclampsia
Aspirin induced lipoxins: alternative to modulate proinflammatory processes in preeclampsia
Biol. Manuela Velásquez Berrio, Biol. MSc. Aura María Gil-Villa, MD. MSc. DSc. Angela Patricia Cadavid Jaramillo
Universidad de Antioquia, Medellín, Colombia.
RESUMEN
La preeclampsia es un síndrome hipertensivo que se presenta a partir de la semana 20 de gestación. El objetivo de este trabajo es describir la producción y los mecanismos de acción de las lipoxinas inducidas por la aspirina y proponerlas como una alternativa adecuada para modular los procesos oxidativos característicos de la preeclampsia y los ciclos proinflamatorios que inician con la cascada de activación del factor nuclear-kappa B, y en consecuencia de sus productos. La preeclampsia se caracteriza por la producción de sustancias proinflamatorias, que inducen la activación de células endoteliales, directa o indirectamente, a través de la activación previa de los monocitos, los cuales pueden generar especies reactivas de oxígeno y expresar moléculas de adhesión que median la interacción con el endotelio, contribuyendo a su estado de disfunción, activación e inducción de la cascada de señalización del factor nuclear-kappa B. La aspirina por su parte, induce la producción de lipoxinas que inhiben la activación del factor nuclear-kappa B mediante el bloqueo de la proteína quinasa IkB, necesaria para desencadenar la activación de la vía canónica y no canónica de este factor nuclear.
Palabras clave: preeclampsia, disfunción endotelial, factor-kappa B, lipoxinas inducidas por la aspirina.
ABSTRACT
Preeclampsia is a hypertensive syndrome that occurs after the 20th weeks of gestation. The objective of this review was to describe the mechanisms of production and action of aspirin- triggered lipoxins in order to consider them as a suitable alternative to modulate oxidative processes, which are characteristic of preeclampsia and proinflammatory cycles starting with cascade activation of nuclear factor-kappa B, consequently of their products. Preeclampsia is characterized by the production of proinflammatory substances that induce directly or indirectly endothelial cell activation,, through prior activation of monocytes, which can generate reactive oxygen species and expression of adhesion molecules that mediate interacting with the endothelium, contributing to its dysfunction, activation and induction of signaling cascade nuclear factor-kappa B. Aspirin induces lipoxin, which inhibits the activation of nuclear factor-kappa B by blocking IkB protein kinase, necessary to trigger the activation of canonical and non-canonical pathway of this nuclear factor.
Keywords: preeclampsia, endothelial dysfunction, nuclear factor-kappa B, aspirin-triggered lipoxins.
INTRODUCCIÓN
La preeclampsia se caracteriza por el aumento de la presión arterial y de los niveles de proteína en orina después de la semana 20 de gestación; es la causa del 2-8 % de las complicaciones que se presentan durante el embarazo, provocando aproximadamente el 40 % de los partos prematuros iatrogénicos.1 El factor de riesgo principal para desarrollar preeclampsia es la primiparidad en el 75 % de los casos y además se asocia con algunos síndromes o trastornos como: diabetes mellitus, enfermedades vasculares antes de la concepción, desórdenes autoinmunes, obesidad y predisposición genética para desarrollar la enfermedad.2 Los efectos negativos de la preeclampsia no se presentan únicamente en la madre, ya que en el feto también se pueden producir alteraciones en el desarrollo del sistema nervioso central, trombocitopenia, prematurez y bajo peso al nacer, lo cual es considerado el factor de riesgo mayor para inducir la muerte fetal en nacimientos provenientes de una preeclampsia severa.3
La aspirina (ASA) a bajas dosis ha sido empleada en la prevención de diversas alteraciones de la gestación, como pérdida gestacional recurrente y preeclampsia.4-6 En una revisión sistemática que involucró 59 ensayos clínicos (37 560 mujeres), se evaluaron diferentes terapias antiplaquetarias para la prevención de la preeclampsia, demostrándose que las bajas dosis de ASA reducían el riesgo de desarrollar este síndrome en aproximadamente el 17 % de los casos.7 A pesar de existir controversia acerca de la efectividad y la administración de este fármaco en el tratamiento de mujeres gestantes, ASA ha sido utilizada desde la antigüedad para tratar múltiples condiciones inflamatorias con resultados satisfactorios y en la actualidad continúa siendo usada en la prevención primaria de enfermedades coronarias y cerebrovasculares.8
El mecanismo de acción tradicional de la aspirina es inhibir irreversiblemente la enzima ciclooxigenasa-1 (COX-1), la cual convierte el ácido araquidónico a endoperóxidos de prostaglandina, y de esta manera, reduce la biosíntesis de prostaglandinas (PG) y de tromboxanos, sin embargo, este evento no explica completamente el repertorio de acciones antiinflamatorias de este medicamento.9 Recientemente, se describió un mecanismo que involucra la biosíntesis transcelular de lipoxinas inducidas por la aspirina, también llamadas epi-lipoxinas o ATL (aspirin-triggered lipoxin) a partir del ácido araquidónico: la aspirina acetila la COX-2 y redirecciona la actividad catalítica de la enzima para producir el ácido 15(R)-hidroxieicosatetraenoico, que a través de la enzima 5-lipoxigenasa es convertido a ATL, la cual es rápidamente liberada.10,11
ATL conduce a la resolución de la inflamación y es un componente angiogénico e inmunomodulador, el cual podría ser prometedor en el tratamiento de la preeclampsia, ya que bloquea la generación de especies reactivas de oxígeno y es un importante antiinflamatorio al reducir la adhesión de polimorfonucleares a células endoteliales estimuladas con el plasma de mujeres con preeclampsia; inhibe la secreción del factor de necrosis tumoral alfa (TNF) y la activación del factor nuclear- kappa B (NF-kB).12-14 El objetivo de este estudio fue mostrar de manera descriptiva los mecanismos de acción y producción de las ATL para proponerlas como una alternativa adecuada para modular el estado oxidativo y proinflamatorio característico en la preeclampsia que se genera a partir de la activación del NF-kB y de sus productos.
DESARROLLO
Activación y disfunción endotelial
En el proceso normal de la placentación temprana, el citotrofoblasto extravelloso fetal invade las arterias espirales uterinas y transforma los pequeños vasos de alta resistencia, en vasos de mayor capacitancia, capaces de proporcionar una perfusión placentaria adecuada para mantener el crecimiento fetal.15 En la preeclampsia, este remodelamiento vascular es incompleto y se produce un flujo arterial útero-placentario reducido con episodios de hipoxia-reperfusión generadores de especies reactivas de oxígeno (ERO), que en última instancia, inducen la liberación de citocinas proinflamatorias, peróxidos lipídicos, y microfragmentos desde la placenta hacia la circulación materna.16 Estos productos pueden estimular una respuesta inflamatoria en el endotelio materno, generada directa o indirectamente, por otras células activadas previamente como los monocitos.17,18 De esta manera, la preeclampsia está asociada con una exagerada activación endotelial e inflamación generalizada en comparación con el embarazo normal.19
En concordancia con otros autores, estos resultados han evidenciado un incremento de productos de la lipoperoxidación lipídica como 8-isoprostano y sustancias reactivas al ácido tiobarbitúrico en el plasma de mujeres con preeclampsia, comparado con los encontrados en el plasma de gestantes normotensas y no gestantes, confirmando un mayor estado de estrés oxidativo presente en la preeclampsia.12,20 Las ERO son sustancias que aumentan la inflamación, acentúan la gravedad de las pacientes con preeclampsia, alteran la relación tromboxano/prostaciclina y la producción de TNF.21
De otro lado, durante la preeclampsia los monocitos/macrófagos, las células asesinas naturales y los linfocitos T activados producen interferón gamma que junto con el TNF y la interleuquina-1b (IL-1b), inhiben la angiogénesis en células endoteliales y desencadenan apoptosis en las células del trofoblasto, que se encargan de la adherencia e implantación del embrión.22
Activación de monocitos
Los productos generados en la preeclampsia, inducen una respuesta inflamatoria que puede iniciarse a partir de la estimulación de los monocitos que al interactuar con el endotelio materno, ocasionan su activación y disfunción.17,18 Lo anterior se ha evidenciado en ensayos in vitro, en los cuales se ha evaluado la producción de ERO y la expresión de la molécula de adhesión intercelular-1 (ICAM-1) en monocitos y en células endoteliales de vena de cordón umbilical humano (HUVEC) cultivadas en presencia del plasma de mujeres con preeclampsia, donde se encontró que los monocitos incrementaban la producción de ERO y la expresión de ICAM -1 tanto en monocultivo como en el cocultivo con las células HUVEC, comparado a su estado basal; en contraste, las células HUVEC aumentaron la producción de ERO y la expresión de ICAM-1 únicamente en cocultivo con los monocitos, sugiriendo que los monocitos son estimulados en un primer lugar y posteriormente inducen la activación de las células endoteliales, favoreciendo el estado proinflamatorio característico de la preeclampsia.18
Mediante el desarrollo de otros modelos in vitro de cocultivo de monocitos y células endoteliales, se ha podido determinar que el plasma de mujeres con preeclampsia potencia la adhesión de estos dos tipos de células después de 16 horas de cultivo en comparación con el efecto del plasma de mujeres gestantes normotensas.23 Por otro lado, en monocitos de sangre periférica de mujeres con preeclampsia se encontró mediante citometría de flujo, la expresión elevada de la integrina CD11b (molécula que media la adhesión de los monocitos al endotelio) y una alta producción basal de ERO en comparación con los monocitos de sangre periférica de mujeres gestantes normotensas, evidenciando la activación característica de estas células durante la preeclampsia.24
Activación del factor nuclear-kappa B
La activación de monocitos y células endoteliales es el resultado de mecanismos moleculares que involucran a diversas vías de señalización como las implicadas en la activación del NF-kB en la preeclampsia, el cual induce un estado inflamatorio por la producción de IL-1b y TNF.16,25 En monocitos aislados a partir de sangre periférica de mujeres con preeclampsia se encontró un incremento del NF-kB activado en comparación con gestantes normotensas.25 Por su parte, en células endoteliales se pudo evidenciar que la mayor concentración de lípidos peroxidados presente en el plasma de las mujeres con preeclampsia, inducían la activación del NF-kB, y que a su vez desencadenaba un aumento en la expresión de ICAM -1.26
El NF-kB está constituido por cinco proteínas: RelA (p65), RelB, Rel (c- Rel), p50, p52; su forma inactiva se une a inhibidores IkB (IkBa, IkBb, IkBz, IkBe, IkBs, Bcl-3, p100 y p105) que impiden su translocación al núcleo mediante la presencia de secuencias de localización nuclear, manteniendo al factor NF-kB en el citoplasma.27 La activación del factor NF-kB puede presentarse por dos vías diferentes: canónica y no canónica, dependiendo del estímulo que inicie la cascada celular.28
La vía canónica puede ser activada por la unión de la IL-1b al receptor 1 de la interleucina 1 (ILR1) y del TNF al receptor 1 del factor de necrosis tumoral (TNFR1); estos últimos inducen el reclutamiento del receptor de TNF asociado al factor 2 (TRAF2), que posteriormente activa a la familia de cinasas de los inhibidores de NF-kB (IKK) constituida por IKKa e IKKb, los cuales actúan uniéndose a IKKg o al modulador esencial de NF-kB (NEMO), formando el complejo NEMO/IKKa/b.29 Este complejo heterodimérico de IKK fosforila a las proteínas IkBb, IkBa y Bcl-3 induciendo su degradación en el proteosoma y posterior liberación de los factores p50 y p65, que se translocan al núcleo e inducen la expresión de los genes necesarios para la producción de citocinas proinflamatorias y moléculas de adhesión como ICAM-1 y la molécula de adhesión vascular (VCAM).30
La vía no canónica puede ser inducida por la presencia de linfotoxina b, ERO o la unión del receptor CD40 a su ligando; cada uno de estos factores tiene la capacidad de inducir el reclutamiento de los receptores de TNF asociados a TRAF2 y TRAF3, los cuales activan la proteína cinasa inductora de NF-kB (NIK) y promueven la acción de IKKa/b, que fosforila a p100 dejando libre al factor RelB. Este último, se une a los factores p50 y p52 para translocarse al núcleo y promover la activación de genes para la expresión de ICAM-1, VCAM, IL-12 e IL-1b (Fig. 1).2
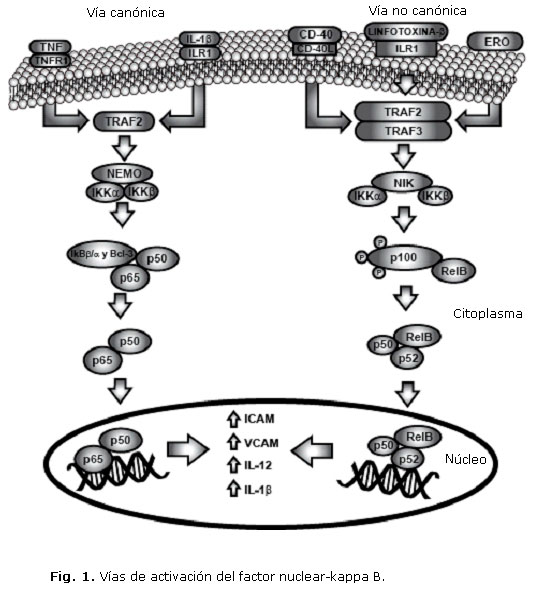
Finalmente, se ha evidenciado que en la decidua y en el plasma de mujeres con preeclampsia existe un aumento en la producción de citocinas proinflamatorias y, una mayor expresión del receptor CD40 y su ligando en monocitos de sangre periférica en comparación con mujeres normogestantes.31 Lo anterior contribuye a favorecer el curso de la inflamación y de la disfunción endotelial, considerando que se estaría induciendo la activación del NF-kB por la vía canónica y la no canónica.32
Lipoxinas inducidas por la aspirina:
1. Síntesis de eicosanoides
La liberación del ácido araquidónico a partir de los fosfolípidos de las membranas celulares es el paso inicial para que se lleve a cabo la síntesis de eicosanoides.33 Esta se produce como respuesta a diversos estímulos físicos, químicos o mecánicos como impulsos nerviosos, antígenos, citocinas, mitógenos, factores tumorales, estrés oxidativo, daño celular, isquemia, hormonas, neuropéptidos, entre otros.33 El metabolismo de los eicosanoides es inmediato y se lleva a cabo a través de tres sistemas enzimáticos principales: a) la ciclooxigenasa (COX), a partir de la cual se originan los prostanoides como las PG, los tromboxanos (TX) y la prostaciclina; b) lipoxigenasas (LOX), de donde se derivan los leucotrienos y las lipoxinas, y c) citocromo P-450, que origina los denominados productos de la vía de las epoxigenasas.34-36
a) Vía de la ciclooxigenasa. Existen principalmente dos isoformas de la enzima, COX-1 y COX-2. La COX-1 es una enzima constitutiva encargada de la síntesis de PG implicadas en la homeostasis general y por consiguiente se expresa en la mayoría de los tejidos del organismo.36 El grado de expresión basal de la enzima varía entre diferentes poblaciones celulares; por ejemplo, el endotelio vascular y las plaquetas se caracterizan por tener concentraciones altas de COX-1.36 Por su parte, la COX-2 es una isoforma no constitutiva en la mayoría de los tejidos y produce prostanoides en los sitios de inflamación, sin embargo, existen algunas excepciones, encontrándose concentraciones altas de la enzima COX-2 en próstata, timo de recién nacido y cerebro.37
La COX se encarga primero de la ciclación y de la oxigenación del ácido araquidónico, formando la PGG2 y después reduce esta última para formar PGH2, a partir de la cual las endoperóxido isomerasas sintetizan PGE2 y PGD2, mientras que por reducción se origina prostaglandina F2a.36 Las PGA2, PGB2 y PGC2, todas derivadas de la PGE2, no se producen en condiciones fisiológicas y se suponen que son productos de la síntesis química. A partir de la PGH2 también se producen otros compuestos inestables y potentes como el TXA2, el cual se forma por acción de la tromboxano sintetasa y se metaboliza rápidamente y de forma no enzimática a TXB2, que es más estable desde el punto de vista químico pero biológicamente menos potente. Finalmente, la prostaciclina o PGI2 es producto de la acción de la prostaciclina sintetasa y también se hidroliza rápidamente en un metabolito inactivo, la 6-ceto-PGF-1a.36
b) Vía de la lipoxigenasa. Las LOX son enzimas citosólicas encargadas de oxidar (sin ciclar) los ácidos grasos poliénicos en el carbono 5 (5-LOX), 12 (12-LOX) o 15 (15-LOX), formando los correspondientes hidroperóxidos lipídicos: HPETE.35 La 5-LOX es la más importante y se localiza principalmente en las células que participan en la respuesta inflamatoria, como neutrófilos, monocitos macrófagos, eosinófilos y mastocitos. Cuando la 5-LOX se une a la proteína activadora FLAP, convierte el ácido araquidónico en ácido 5-hidroperoxi-6,8,11,14 eicosatetraenoico (5-HPETE), el cual puede ser transformado en ácido 5-hidroxi-6,8,11,14 eicosatetraenoico (5-HETE) o en el epóxido 5,6 conocido como leucotrieno A4 (LTA4). El LTA4 por acción de la leucotrieno A hidrolasa se convierte en LTB4 o al conjugarse con glutatión mediante la glutatión S transferasa, origina el LTC4. El LTD4 se forma al separarse el ácido glutámico del LTC4, y el LTE4 como consecuencia de la posterior pérdida de glicina. La recuperación siguiente del ácido glutámico da lugar al LTF4. Al igual que ocurre con las PG, las diferentes poblaciones celulares son capaces de producir leucotrienos distintos, por ejemplo los neutrófilos producen principalemte LTB4.37
A partir del ácido araquidónico, la 12-LOX produce 12-HPETE, mientras que la 15-LOX forma 15-HPETE, y por acción posterior de una peroxidasa o de forma no enzimática, se originan los correspondientes 12-HETE y 15-HETE. La acción de una hidroperoxidasa de ácidos grasos sobre la 12-HPETE transforma este componente en hepoxilinas A y B, las cuales pueden llegar a ser metabolizadas y transformadas en trioxilinas. De modo similar, las lipoxinas, sintetizadas predominantemente en los neutrófilos, son producto de la acción combinada (biosíntesis transcelular) de 15- y 5-LOX que originan un compuesto inestable, el 5(6)-epóxido-tetraeno que, posteriormente puede convertirse en lipoxina A4 o lipoxina B4 mediante procesos enzimáticos diferentes.37
c) Vía de la epoxigenasa o del citocromo P-450. La acción del citocromo P-450 sobre el ácido araquidónico produce un conjunto variado de ácidos epoxieicosatrienoicos (EET) e isómeros de los ácidos hidroxieicosatetraenoicos (D-HETE).34
2. Papel de la aspirina sobre la síntesis de eicosanoides
Debido a que ASA es rápidamente deacetilada a salicilato, se ha asumido que los efectos antiinflamatorios del medicamento son principalmente mediados por el salicilato.38 Sin embargo, es importante resaltar las acciones farmacológicas de ASA determinadas tanto por el metabolito primario del ácido salicílico (salicilato), como por la molécula de acetato.39
a) Salicilato. Posee acciones independientes de la inhibición de la síntesis de PG e interactúa con cascadas de señalización celular que controlan la inflamación, el dolor, la tumorogénesis, y también la apoptosis y sus mediadores como eventos centrales para el daño celular.40
- Desacoplamiento de la fosforilación oxidativa. Existen acciones directas pero no específicas de los salicilatos sobre las membranas celulares, predominando la acción de estos componentes como protonóforos en las membranas mitocondriales, lo cual ocasiona el desacoplamiento de la fosforilación oxidativa.41 Este evento produce un aumento en el consumo de oxígeno y en la producción de dióxido de carbono. Adicional a la estimulación de la respiración que ejercen los salicilatos, lo anterior es una de las razones por las cuales estos componentes pueden ocasionar hiperventilación. De otro lado, el desacoplamiento de la fosforilación oxidativa disminuye la formación de trifosfato de adenosina o ATP y como consecuencia induce la liberación de adenosina a los fluidos extracelulares, lo cual al parecer, contribuye a los efectos antiinflamatorios del salicilato.41
- Acción sobre las proteínas cinasas. Las altas concentraciones de salicilatos interfieren con las cascadas de señalización en las cuales están involucradas las proteínas quinasas, que incluyen las proteínas cinasas activadas por mitógeno o MAPK cinasas. Exceptuando la activación de p38 MAPK inducida por salicilatos, los efectos descritos son en su mayoría inhibidores. Esta podría ser una de las razones por las cuales se han observado efectos inhibidores de los salicilatos "corriente abajo" de las cinasas sobre varios factores de transcripción nucleares.42
- Interacción entre los salicilatos y NF-kB. La inhibición de la cascada de activación del factor NF-kB es uno de los mecanismos antiinflamatorios de ASA, que dependen de la concentración utilizada y del tiempo de incubación previo. En ensayos in vitro se han implementado dosis de ASA que actúan desde los primeros 30 min, utilizadas en un rango de concentración de 0.1 a 20 milimolar en células epiteliales.43 ASA impide la acción de la proteína cinasa IKK en células estimuladas con IL-1b y TNF, imposibilitando la expresión de genes involucrados en la respuesta proinflamatoria, mediante el bloqueo de la fosforilación de las proteínas inhibidoras IkB.44 Sin embargo, otros autores que incluyen a Saunders y otros, han sugerido que las acciones farmacológicas de ASA y de los salicilatos son mediados por la inhibición de la unión y de la transactivación del factor de transcripción C/EBP (CCAAT /enhancer binding protein, por sus siglas en inglés), más que por la interferencia que pueden tener estos medicamentos con NF-kB.40
- Transcripción de las enzimas óxido nítrico sintasa inducible (iNOS) y COX-2: La transcripción de iNOS en respuesta a la estimulación con LPS e interferón gama también requiere la unión de C/EBP a la secuencia CCAAT en el promotor del gen iNOS. Por su parte, el grupo de Wu KK y otros, encontraron que tanto el ARN mensajero como la proteína para iNOS fueron suprimidos por la aspirina y el salicilato de sodio, de manera similar a la supresión ejercida sobre el promotor de COX-2.45
b) Molécula de acetato. Tiene la capacidad de transacetilar proteínas de forma no selectiva y no enzimática, lo cual es un mecanismo postranscripcional crucial para el mantenimiento de las funciones celulares.
- Inhibición de actividad enzimática. ASA inhibe irreversiblemente la actividad enzimática de COX, actuando sobre ambas isoformas dependiendo de la dosis: a dosis altas inhibe la COX-2, y tanto a bajas como a altas dosis inhibe la COX-1, uniéndose a ellas de forma irreversible realizando un proceso de acetilación. En la COX-1 acetila la serina en la posición 530 evitando así, la unión del ácido araquidónico y, en la COX-2, acetila la serina en la posición 516 que es el sitio activo homólogo al de la enzima COX-1, pero no impide que pueda unirse el ácido araquidónico, sino que evita que la COX-2 pueda sintetizar PG.46
- Generación de nuevos productos. Teniendo en cuenta lo anterior, la COX-2 queda habilitada para producir ácido 15(R)-HETE, un producto que muestra una estereoquímica invertida en el carbono 15, comparada a los productos de la enzima LOX (configuración S). Posteriormente, mediante biosíntesis transcelular con la enzima 5-LOX leucocitaria y a partir del 15(R)-HETE, se da lugar a las R-lipoxinas, que poseen funciones antiinflamatorias y de resolución de la inflamación y en consecuencia, potencian el efecto de ASA (Fig. 2).47

- Inhibición de factores de transcripción. Se ha descrito que las ATL inhiben la activación del factor NF-kB bloqueando la acción de IKK a una concentración de 100 y 500 nanomolar, lo cual impide la fosforilación de proteínas inhibidoras del factor nuclear, la producción de anión superóxido y de TNF, actuando mediante la unión a su receptor ALX o CysLT1, expresados en células endoteliales, monocitos, enterocitos y polimorfonucleares. 14,48
- Acetilación de otros blancos celulares. ASA regula positivamente la formación de óxido nítrico a partir de la enzima óxido nítrico sintasa endógena o eNOS, lo cual no solamente mejora la perfusión local sino que incrementa la expresión de dos proteínas de protección endotelial denominadas hemo oxigenasa 1 y ferritina. 49 Al aumentar la concentración de hemo oxigenasa, incrementa subsiguientemente la formación de bilirrubina, monóxido de carbono y ferritina, a la cual se le han encontrado propiedades antioxidantes. Lo anterior, ha sido considerado una importante acción vasoprotectora de ASA independiente de la inhibición de PG.
Considerando que ASA se une de forma irreversible tanto a la COX-1 como a la COX-2, se requiere que estas enzimas sean resintetizadas por la célula; esta síntesis no puede ser realizada por las plaquetas, ya que son células que no poseen núcleo y por ende carecen de los medios necesarios para sintetizar proteínas, por lo que las plaquetas afectadas con ASA deben ser reemplazadas en el organismo por otras sin COX bloqueada. Lo anterior permite que el efecto antiagregante de ASA sea más duradero que los otros antiinflamatorios que se unen de forma reversible a la COX; la duración del efecto sobre las plaquetas dura tanto como el período de vida de estas, que va de 8 a 10 días.50
CONCLUSIONES
El estado de hipoxia reperfusión, el remodelamiento vascular incompleto y el flujo arterial útero placentario reducido, característico de la preeclampsia conducen en última instancia, a la activación del endotelio vascular directamente, o por medio de otras células activadas como los monocitos que en consecuencia expresan moléculas de adhesión, producen lípidos peroxidados, citocinas proinflamatorias y ERO. Estas sustancias inducen la activación del factor NF-kB por la vía canónica y no canónica simultáneamente, promoviendo el curso de la respuesta inflamatoria que, finalmente, contribuye a mantener los síntomas propios de la preeclampsia. De otro lado, las lipoxinas inducidas por la aspirina o ATL, poseen la capacidad de modular la activación del factor NF-kB en células endoteliales en concentraciones más bajas a las utilizadas con ASA, lo que demuestra que estas podrían ser más potentes y adecuadas para modular la generación de moléculas producto de la activación del NF-kB; sin embargo, a pesar de conocerse las propiedades de las ATL, no existen reportes en la literatura de la utilización de estos mediadores lipídicos en el contexto reproductivo, específicamente en la preeclampsia.
Agradecimientos
Apoyo financiero Estrategia Sostenibilidad Universidad de Antioquia 2013-2014. Aura M. Gil-Villa es becaria de COLCIENCIAS.
REFERENCIAS BIBLIOGRÁFICAS
1. Meis PJ, Goldenberg RL, Mercer BM. The preterm prediction study: risk factors for indicated preterm births. Maternal-Fetal Medicine Units Network of the National Institute of Child Health and Human Development. Am J Obstet Gynecol. 1998;178:562-7.
2. Arngrimsson R, Bjornsson S, Geirsson RT, Bjornsson H, Walker JJ, Snaedal G. Genetic and familial predisposition to eclampsia and pre-eclampsia in a defined population. Br J Obstet Gynaecol. 1990;97:762-9.
3. Simpson LL. Maternal medical disease: risk of antepartum fetal death. Semin Perinatol. 2002;26:42-50.
4. Heyborne KD. Preeclampsia prevention: lessons from the low-dose aspirin therapy trials. Am J Obstet Gynecol. 2000;183:523-8.
5. Ogasawara MS, Iinuma Y, Aoki K, Katano K, Ozaki Y, Suzumori K. Low-dose aspirin is effective for treatment of recurrent miscarriage in patients with decreased coagulation factor XII. Fertil Steril. 2001;76:203-4.
6. Roberge S, Giguere Y, Villa P. Early administration of low-dose aspirin for the prevention of severe and mild preeclampsia: a systematic review and meta-analysis. Am J Perinatol. 2012;29:551-6.
7. Duley L, Henderson-Smart DJ, Meher S, King JF. Antiplatelet agents for preventing pre-eclampsia and its complications. Cochrane Database Syst Rev. 2007:CD004659.
8. Berger JS, Roncaglioni MC, Avanzini F, Pangrazzi I, Tognoni G, Brown DL. Aspirin for the primary prevention of cardiovascular events in women and men: a sex-specific meta-analysis of randomized controlled trials. JAMA. 2006;295:306-13.
9. Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol. 1971;231:232-5.
10. Claria J, Serhan CN. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc Natl Acad Sci US A. 1995;92:9475-9.
11. Serhan CN, Yacoubian S, Yang R. Anti-inflammatory and proresolving lipid mediators. Annu Rev Pathol. 2008;3:279-312.
12. Gil-Villa AM, Norling LV, Serhan CN, Cordero D, Rojas M, Cadavid A. Aspirin triggered-lipoxin A4 reduces the adhesion of human polymorphonuclear neutrophils to endothelial cells initiated by preeclamptic plasma. Prostaglandins Leukot Essent Fatty Acids. 2012;87:127-34.
13. Ariel A, Chiang N, Arita M, Petasis NA, Serhan CN. Aspirin-triggered lipoxin A4 and B4 analogs block extracellular signal-regulated kinase-dependent TNF-alpha secretion from human T cells. J Immunol. 2003;170:6266-72.
14. Jozsef L, Zouki C, Petasis NA, Serhan CN, Filep JG. Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 inhibit peroxynitrite formation, NF-kappa B and AP-1 activation, and IL-8 gene expression in human leukocytes. Proc Natl Acad Sci U S A. 2002;99:13266-71.
15. Lam C, Lim KH, Karumanchi SA. Circulating angiogenic factors in the pathogenesis and prediction of preeclampsia. Hypertension. 2005;46:1077-85.
16. Redman CW, Sargent IL. Placental debris, oxidative stress and pre-eclampsia. Placenta. 2000;21:597-602.
17. Hou L, Zhu Y, Ma X, Li J, Zhang W. Serum protein microarray analysis of patients with preeclampsia. Mol Med Report. 2012;6:83-7.
18. Faas MM, van Pampus MG, Anninga ZA. Plasma from preeclamptic women activates endothelial cells via monocyte activation in vitro. J Reprod Immunol. 2010;87:28-38.
19. Redman CW, Sacks GP, Sargent IL. Preeclampsia: an excessive maternal inflammatory response to pregnancy. Am J Obstet Gynecol. 1999;180:499-506.
20. Mikic AN, Brkic S, Maric D, Sekulic B, Cetkovic A, Mitic G. Thiobarbituric acid reactive substances as marker of oxidative stress in pregnancies with pre-eclampsia. Med Pregl. 2011;64:377-80.
21. Serdar Z, Gur E, Colakoethullary M, Develioethlu O, Sarandol E. Lipid and protein oxidation and antioxidant function in women with mild and severe preeclampsia. Arch Gynecol Obstet. 2003;268:19-25.
22. Peracoli MT, Bannwart CF, Cristofalo R. Increased reactive oxygen species and tumor necrosis factor-alpha production by monocytes are associated with elevated levels of uric acid in pre-eclamptic women. Am J Reprod Immunol. 2011;66:460-7.
23. Flood-Nichols SK, Stallings JD, Gotkin JL, Tinnemore D, Napolitano PG, Ippolito DL. Elevated ratio of maternal plasma ApoCIII to ApoCII in preeclampsia. Reprod Sci. 2011;18:493-502.
24. Gervasi MT, Chaiworapongsa T, Naccasha N. Phenotypic and metabolic characteristics of maternal monocytes and granulocytes in preterm labor with intact membranes. Am J Obstet Gynecol. 2001;185:1124-9.
25. Luppi P, Tse H, Lain KY, Markovic N, Piganelli JD, DeLoia JA. Preeclampsia activates circulating immune cells with engagement of the NF-kappaB pathway. Am J Reprod Immunol. 2006;56:135-44.
26. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21:103-15.
27. Jacobs MD, Harrison SC. Structure of an IkappaBalpha/NF-kappaB complex. Cell. 1998;95:749-58.
28. Gringhuis SI, den Dunnen J, Litjens M. Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-kappaB activation through Raf-1 and Syk. Nat Immunol. 2009;10:203-13.
29. Verma UN, Yamamoto Y, Prajapati S, Gaynor RB. Nuclear role of I kappa B Kinase-gamma/NF-kappa B essential modulator (IKK gamma/NEMO) in NF-kappa B-dependent gene expression. J Biol Chem. 2004;279:3509-15.
30. Urso C, Caimi G. Oxidative stress and endothelial dysfunction. Minerva Med. 2011;102:59-77.
31. Hayman R, Brockelsby J, Kenny L, Baker P. Preeclampsia: the endothelium, circulating factor(s) and vascular endothelial growth factor. J Soc Gynecol Investig. 1999;6:3-10.
32. Lockwood CJ, Huang SJ, Krikun G. Decidual hemostasis, inflammation, and angiogenesis in pre-eclampsia. Semin Thromb Hemost. 2011;37:158-64.
33. Astudillo AM, Balgoma D, Balboa MA, Balsinde J. Dynamics of arachidonic acid mobilization by inflammatory cells. Biochimica et biophysica acta. 2012;1821:249-56.
34. Fleming I. Cytochrome P450-dependent eicosanoid production and crosstalk. Curr Opin Lipidol. 2011;22:403-9.
35. Haeggstrom JZ, Funk CD. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem Rev. 2011;111:5866-98.
36. Smith WL, Urade Y, Jakobsson PJ. Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem Rev. 2011;111:5821-65.
37. Masferrer JL, Zweifel BS, Manning PT. Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:3228-32.
38. Amann R, Peskar BA. Anti-inflammatory effects of aspirin and sodium salicylate. Eur J Pharmacol. 2002;447:1-9.
39. Vane JR, Botting RM. The mechanism of action of aspirin. Thromb Res. 2003;110:255-8.
40. Saunders MA, Sansores-Garcia L, Gilroy DW, Wu KK. Selective suppression of CCAAT/enhancer-binding protein beta binding and cyclooxygenase-2 promoter activity by sodium salicylate in quiescent human fibroblasts. J Biol Chem. 2001;276:18897-904.
41. Yoshida Y, Singh I, Darby CP. Effect of salicylic acid and calcium on mitochondrial functions. Acta Neurol Scand. 1992;85:191-6.
42. Schwenger P, Bellosta P, Vietor I, Basilico C, Skolnik EY, Vilcek J. Sodium salicylate induces apoptosis via p38 mitogen-activated protein kinase but inhibits tumor necrosis factor-induced c-Jun N-terminal kinase/stress-activated protein kinase activation. Proc Natl Acad Sci U S A. 1997;94:2869-73.
43. Yan F, Polk DB. Aminosalicylic acid inhibits IkappaB kinase alpha phosphorylation of IkappaBalpha in mouse intestinal epithelial cells. J Biol Chem. 1999;274:36631-6.
44. Yoo CG, Lee S, Lee CT, Kim YW, Han SK, Shim YS. Effect of acetylsalicylic acid on endogenous I kappa B kinase activity in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2001;280:L3-9.
45. Cieslik K, Zhu Y, Wu KK. Salicylate suppresses macrophage nitric-oxide synthase-2 and cyclo-oxygenase-2 expression by inhibiting CCAAT/enhancer-binding protein-beta binding via a common signaling pathway. J Biol Chem. 2002;277:49304-10.
46. DeWitt DL, el-Harith EA, Kraemer SA. The aspirin and heme-binding sites of ovine and murine prostaglandin endoperoxide synthases. J Biol Chem. 1990;265:5192-8.
47. Lecomte M, Laneuville O, Ji C, DeWitt DL, Smith WL. Acetylation of human prostaglandin endoperoxide synthase-2 (cyclooxygenase-2) by aspirin. J Biol Chem. 1994;269:13207-15.
48. Wang YP, Wu Y, Li LY. Aspirin-triggered lipoxin A4 attenuates LPS-induced pro-inflammatory responses by inhibiting activation of NF-kappaB and MAPKs in BV-2 microglial cells. J Neuroinflammation. 2011;8:95.
49. Grosser N, Abate A, Oberle S. Heme oxygenase-1 induction may explain the antioxidant profile of aspirin. Biochem Biophys Res Commun. 2003;308:956-60.
50. Chiang N, Arita M, Serhan CN. Anti-inflammatory circuitry: lipoxin, aspirin-triggered lipoxins and their receptor ALX. Prostaglandins Leukot Essent Fatty Acids. 2005;73:163-77.
Recibido: 9 de junio de 2013.
Aprobado: 24 de junio de 2013.
Manuela Velásquez Berrío. Laboratorio 534, Sede de investigación universitaria (SIU). Universidad de Antioquia. Carrera 53 # 61-30. Medellín, Colombia. Teléfonos: (57-4) 219 64 76 219 64 77. Correo electrónico:manue260789@hotmail.com; reproduccion@medicina.udea.edu.co

