










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Obstetricia y Ginecología
versión On-line ISSN 1561-3062
Rev Cubana Obstet Ginecol vol.41 no.1 Ciudad de la Habana ene.-mar. 2015
OBSTETRICIA
Reordenamientos cromosómicos estructurales en el diagnóstico citogenético prenatal y postnatal acorde a su origen
Structural chromosomal rearrangements in prenatal and postnatal cytogenetic diagnosis according to their origin
MSc. Olga Luisa Quiñones Maza,I DrC. Luis Alberto Méndez Rosado,II Dr. Jorge Quintana Aguilar,III Téc. Ursulina Suárez Mayedo,II Lic. Minerva García Rodríguez,III Anduriña Barrios Martínez II, MSc. Nereida González García,II MSc. Michel Soriano Torres,II MSc. Enny Morales Rodríguez,II Lic. Marylin del Sol GonzálezII
I Centro Provincial de Genética Médica. La Habana, Cuba.
II Centro Nacional de Genética Médica. La Habana, Cuba.
III Servicio Municipal de Genética Médica. Municipio Playa. La Habana, Cuba.
RESUMEN
Introducción: según el origen, existen diferentes tipos de reordenamientos cromosómicos estructurales tales como: heredados, de “novo” y desconocidos; en dependencia de esto será el riesgo en la descendencia y el asesoramiento genético específico a los pacientes y a sus familiares.
Objetivo: describir los diferentes reordenamientos cromosómicos estructurales, detectados según su origen.
Métodos: se utilizó la información necesaria contenida en los registros de los laboratorios de Citogenética de los Centros de Genética Médica Nacional y Provincial de La Habana. Se confeccionó una base de datos en Excel. Se identificaron 249 portadores de reordenamientos cromosómicos estructurales de 23 403 casos diagnosticados. Se identificó el origen de los mismos con datos de los registros, basados en la extensión de estudios citogenéticos indicados a padres o familiares que permitieran acreditarlo.
Resultados: el origen de causa desconocida fue el de mayor frecuencia para 48,2 %; predominaron en estudios postnatales con 82,5%. Las heredadas con un 43,0 %, prevalecieron en estudios prenatales (58,0 %); las de origen materno en el 56,0 %. Las heredadas maternas fueron mayores en las inserciones, con valor de 100,0 %, mientras que el origen paterno predominó en las inversiones con valores de 90,0 %.
Conclusiones: el origen de causa desconocida predominó en estudios postnatales, mientras que en los prenatales prevalecieron las de origen heredado. Las heredadas maternas fueron mayoritarias. Es necesario como una estrategia de prevención, incrementar la detección del origen de los RCE en los portadores, así como la extensión del estudio a los miembros de la familia de portadores.
Palabras clave: reordenamientos cromosómicos estructurales, estudios citogenéticos prenatales y postnatales.
ABSTRACT
Introduction: According to the source, there are different types of structural chromosome rearrangements such as inherited, "novo" and unknown; depending on the risk in offspring and specific genetic counseling to patients and their families.
Objective: Describe different structural chromosomal rearrangements detected by origin.
Methods: The necessary information in the records from contained of CytogeneticsNational Laboratory Center and Provincial Medical Genetics Laboratory Centers in Havanawas used. A database in Excel was made. 249 carriers of structural chromosomal rearrangements of 23,403 diagnosed cases were identified. The origin was identified with data records based on the extent of cytogenetic indicated studies that allowed parents or relatives to accredit.
Results:The unknowncause of originwasmore frequent(48.2%); postnatalstudiespredominated (82.5%). The Inheritedcause of originwere present in43.0%, prenatalstudies prevailed (58.0%); thematernaloriginin56.0%. Maternalinheritedcauses were higher inthe inserts, valued at 100.0%, while thepaternal originprevailed ininversions with values of90.0%.
Conclusions: the unknown cause of origin dominates postnatal studies, while the prenatal prevailed in inherited origin. Maternal inherited causes were the majority. As a prevention strategy, it is necessary to increase the detection of the origin of the RCE in carriers, as well as the extension of the study to carriers’ members of the family.
Keywords: structural chromosomal rearrangements, prenatal and postnatal cytogenetic studies.
INTRODUCCIÓN
Existen según su origen, diferentes tipos de reordenamientos estructurales tales como: los heredados o familiares; los de “novo” y los desconocidos.1
Las anomalías estructurales balanceadas o no, pueden ser heredadas de padres portadores o pueden ocurrir como reordenamientos de novo, son formado en un gameto simple o zigoto. Si el reordenamiento estructural es heredado, existe un bajo riesgo de discapacidad física o mental producto del reordenamiento. Cuando la anomalía ocurre como un evento de novo y los padres tienen un cariotipo normal, el riesgo de una enfermedad genética o un fenotipo afectado se ve incrementada, en comparación de si aparece como un reordenamiento balanceado. Esto puede ser el resultado de alguna deleción o duplicación submicroscópicas en los puntos de rupturas, o debido a cambios funcionales de los genes cercanos a los puntos de rupturas o por cambios en las regiones de los genes reguladores.2
Para un portador balanceado (heterozigótico), el problema fenotípico puede ser dificultades en la reproducción evidenciada por infertilidad, abortos espontáneos, descendencia anómala. Estas dificultades provienen de un apareamiento y segregación anómala en la profase de la primera meiosis, cuando ocurre el apareamiento entre cromosomas homólogos.2
Las consecuencias de los portadores de reordenamientos balanceados, si son identificados prenatal, son difíciles de predecir y ofrecer asesoramiento genético específico, cuando se comprueba que su origen es de novo.2
La mayoría de los reordenamientos estructurales desbalanceados aparecen por primera vez (“de novo”), aunque algunos pueden ser heredados de padres portadores de anomalías balanceadas. Algunos portadores de estas anomalías desequilibradas que son compatibles con la vida, son producto de segregaciones anómalas en la meiosis de padres portadores balanceados.3-5
La existencia del Programa Nacional de Detección y Prevención de Enfermedades Cromosómicas, que acumula más de 30 años de trabajo, encabezado por el laboratorio de Citogenética de La Habana, con experiencia e información en este trabajo, y un aval de estudios científicos y publicaciones, ha realizado cientos de diagnósticos de anomalías cromosómicas, en estudios prenatales y postnatales a población de riesgo definida para estas enfermedades.6-12
Dentro de las aberraciones estructurales en el universo estudiado, sería bueno conocer en cuanto al origen de las mismas, cuántas son y cuáles sus características?, esto permitiría conocer, como será el riesgo en la descendencia y el asesoramiento genético específico a los pacientes y a sus familiares.
Este conocimiento permitiría en los casos en que el origen del reordenamiento estructural es de origen heredado, la extensión del estudio a la familia de los portadores de una manera más rápida, fácil y con un menor costo de detectar a nuevos portadores. Esto influiría en las estrategias de prevención y planificación familiar, lo que es posible, a través, de los servicios de genética con que contamos en la actualidad, con el nivel de atención primario del país.11
El presente trabajo pretende contribuir, al conocimiento de los reordenamientos cromosómicos estructurales (RCE) en cuanto al origen de los mismos; a la aplicación en los portadores y sus familiares de mejores estrategias de prevención y mejoras en la calidad, de un asesoramiento genético más específico, así como la posible utilización de técnicas de citogenética molecular en los casos que lo requieran.
Por lo que se propone, describir los diferentes orígenes de los RCE; su relación con los tipos de estudios citogenéticos y de reordenamientos estructurales. Trabajo que hasta el presente no se había realizado en el país, donde se recopila tanto volumen de diagnósticos, en un período mayor de tiempo.
MÉTODOS
Se utilizó la información de los registros de los laboratorios de Citogenética del Centro Nacional de Genética Médica (CNGM) y del Centro Provincial de Genética de Ciudad de La Habana (CPGM) en el período comprendido entre enero de 1984 y diciembre de 2008 (25 años). Del primer laboratorio se recogieron los datos hasta el 2006 y del segundo a partir del mes de julio de 2006 hasta diciembre de 2008.
Los registros contenían la información general necesaria, así como el resultado de los diagnósticos de los estudios cromosómicos realizados a la población de riesgo, que fue remitida fundamental, de Ciudad de La Habana, La Habana, Pinar del Río e Isla de la Juventud, en menor cuantía de Matanzas, Villa Clara, Sancti Spíritus, Camagüey, Ciego de Ávila, Las Tunas, Granma, Holguín y Santiago de Cuba.
Se realizó un análisis retrospectivo de los RCE, en el diagnóstico citogenético. Del total de casos diagnosticados (23 403), sólo se incluyó la información de aquellos, donde se detectó algún RCE como dato primario (249), así como se identificó el origen de los mismos con datos colaterales de los mismos registros, basados en la extensión de estudios citogenéticos indicados a padres o familiares que permitieran acreditar el origen de las anomalías estudiadas.
Para esto se identificaron las variables siguientes (según el origen) y se relacionaron con los tipos de estudios citogenéticos.
Tipos de estudios
- Prenatales: los casos diagnosticados mediante análisis cromosómicos, en muestras de líquido amniótico, por lo general entre las 16 y 20 semanas de gestación.
- Postnatales: los casos diagnosticados mediante estudios cromosómicos en sangre periférica.
Origen
Heredado: los casos en los cuales quedó demostrado que fue trasmitido por uno de los padres.
Materno o paterno : si el reordenamiento fue heredado de la madre o del padre.
“De novo” : los casos en los cuales quedó demostrado que apareció por primera vez en el portador, los padres tuvieron cariotipos normales.
Desconocido : los casos en los cuales no se pudo demostrar el origen.
Se excluyeron las variantes cromosómicas normales como las inversiones de los cromosomas 9 y Y los polimorfismos de la heterocromatina de los cromosomas 1 y 16. Para el procesamiento de los resultados se utilizó, la estadística descriptiva con porcientos y proporciones.
Aspectos éticos
Para la realización de este trabajo no se utilizaron datos personales de los pacientes. Se cumplió, estricto, el principio de la bioética sobre la confidencialidad y seguridad de los resultados médicos.
RESULTADOS
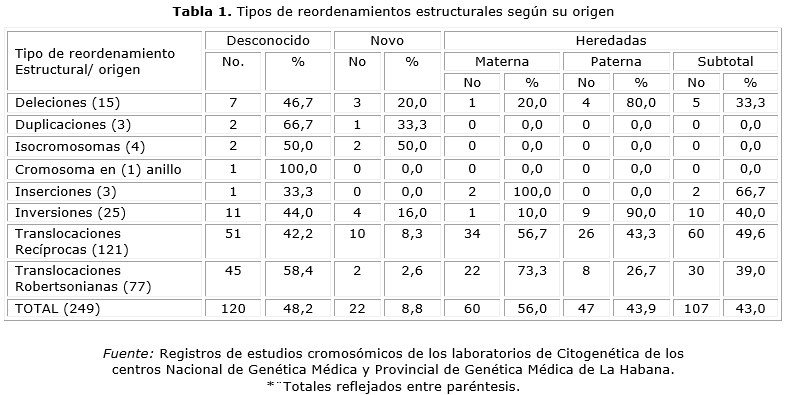
Durante los 25 años estudiados (1984-2008) se realizaron un total de 23 403 diagnósticos cromosómicos (tabla 1). Se identificaron 249 alteraciones cromosómicas estructurales. Se estimó, que el origen de causa desconocido fue el de mayor frecuencia para un 48,2 %; seguidos por las heredadas con un 43,0 %, dentro de estas, las de origen materno en el 56,0 %; mientras que la de menor frecuencia resultó ser la “de novo” con un 8,8%.
Si analizamos las incidencias generales obtuvimos que para el origen desconocido fue de 1 en 195; seguidas por las heredadas de 1 en 218 y las “de novo” de 1 en 1063. Lo que significa que de cada 1000 pacientes diagnosticados se detectaron 4,6 anomalías estructurales heredadas y cerca de una alteración “de novo”.
Los RCE heredados, se detectaron con las mayores frecuencias en las inserciones, translocaciones recíprocas y las inversiones con valores de 66,7 %, 49,6 % y 40,0 % en orden.
Para las duplicaciones, isocromosoma y cromosoma en anillo, no hubo representatividad en el origen heredado.
Las deleciones, fueron heredadas en el 33,3 %. Según datos originales se conoció que, fueron producidos de segregaciones anómalas de portadores de translocaciones recíprocas en el 60,0 % (3/5); el 20,0 % de inversiones (1/5) y el otro 20,0 % de una deleción (1/5).
La herencia de origen materno, fue mayor en las inserciones, translocaciones Robertsonianas y recíprocas con valores de 100,0 %, 73,3 % y 56,7 % al respecto, mientras que el origen paterno predominó en reordenamientos como las inversiones y deleciones, con valores de 90,0 %, 80,0 % mutuo.
En las alteraciones cromosómicas estructurales “de novo”, se pudo apreciar que las mayores cifras de frecuencia, se obtuvieron en los isocromosomas, duplicaciones y deleciones con el 50,0 %, 33,3 % y 20,0 % equitativo.
Otros indicadores estudiados fueron los relacionados con el tipo de estudio y el origen del reordenamiento estructural como se muestra en la tabla 2. Al comparar la procedencia de los portadores de reordenamientos con los tipos de estudios, se apreció que las de origen desconocido predominaron en estudios postnatales con el 82,5 % (99/120), mientras que en análisis prenatales representaron el 17,5 % (21/120).
La incidencia general de las anomalías estructurales prenatales familiares fue de 1 en 273, esto significó que se detectaron 3,7 por cada 1000 diagnósticos. Esta frecuencia se incrementó en 1,3 veces, con respecto a lo reportado por otros investigadores, en que encontraron frecuencias de 2/1000.13
Del total de portadores de rearreglos cromosómicos heredados o familiares, fueron diagnosticados mediante análisis prenatales el 57,9 % (62/107), mientras que en los postnatales el 42,1 % (45/107).
La incidencia de los reordenamientos estructurales prenatales “de novo” fue de 1 en 941, esto significó que se detectaron 1,1 por cada 1000 diagnósticos. Del total de portadores de anomalías estructurales “de novo”, fueron detectados el 81,8 % (18/22) en estudios prenatales, mientras que en postnatales el 18,2 % (4/22).
La distribución de las anomalías estructurales según el origen de las mismas y los tipos de estudios se muestra en la tabla 3. Se observó que en estudios prenatales, las que más contribuyeron al número de reordenamientos cromosómicos de origen heredado fueron las translocaciones recíprocas y Robertsonianas con valores de 48,4 % (30/62) y 30,6 % (19/62) al respecto; mientras que en las “de novo” fueron las translocaciones recíprocas y las inversiones con frecuencias de 44,4 % (8/18) y 22,2 % (4/18) en orden.
En estudios postnatales, las más representativas del origen heredado fueron: las translocaciones recíprocas y Robertsonianas con cifras de 66,7 % (30/45) y 24,4 % (11/45) en orden; mientras que, en las “de novo” fueron las translocaciones recíprocas y las deleciones que aportaron iguales frecuencias con un 50,0 % (2/4).
DISCUSIÓN
Dentro de los RCE de origen conocido, los heredados son la mayoría y los de menor frecuencia los “de novo”, esto coincide con los resultados obtenido en el presente trabajo y por otros autores.1-3
La incidencia de reordenamientos “de novo” en el presente estudio se incrementó entre 1,3 y 2,6 veces si lo comparamos con los estudios realizados por Milunsky A, que plantea para recién nacidos y amniocentesis valores de incidencias entre 0,4/1000 y 0,7/1000 al respecto. 14
En el estudio las translocaciones y las inversiones heredadas (49,6 % y 40,0 %) están en mayoría. Este comportamiento es bien reportado en la literatura,1,2 los resultados lo confirman. Están presente en general en el 0,6-1 % de los individuos, la mayoría son heredadas y de transmisión familiar a través de generaciones.1,2
En cuanto a los valores de frecuencia en el estudio, las translocaciones recíprocas concuerdan con lo reportado por artículos revisados que, muestran cifras que oscilan entre 40,0 % y 76,0 %.14,15 No ocurre así con las inversiones en que, la frecuencia (40,0 %) es menor a la publicada en el artículo de Forrester M y colaboradores, que alcanza cifras de frecuencia mayores (93,0 %).16 Se considera que haya sido el tamaño de muestra, o que haya incluido inversiones del tipo de variantes cromosómicas normales.
La frecuencia de las deleciones heredadas (33,3 %), en el estudio no concuerda con lo publicado por Forrester M y colaboradores que plantean que, las deleciones heredadas detectadas en su estudio tienen cifras de sólo el 27,8 %,17 es un hallazgo citogenético azaroso haber detectado este porciento de deleciones heredadas. Se tiene en cuenta, según la literatura consultada, que son consideradas como un evento raro, ya que la mayoría de las deleciones en humanos ocurren espontánea en la línea germinal de uno de los padres de un individuo afectado y raro son heredadas producto de una segregación adyacente de una translocación heterocigótica o por la recombinación de una inversión, 3,16,18 como ocurrió en el estudio.
La herencia materna en el estudio se correspondió con la bibliografía consultada que plantea que esta es más frecuente que la de origen paterno.14,15 Sin embargo, en cuanto al origen paterno detectado en las inversiones y deleciones (90,0 %, 80,0 %) no se correspondió con reportes consultados, que plantean que éstas tienen cifras de frecuencia de origen materno de 54,0 % y 60,6 % equitativas.16,17
En cuanto a los RCE “de novo”, los resultados coinciden con estudios realizados por otros autores.15,19 Sin embargo, no se corresponden con lo publicado por Forrester M, en cuanto a las deleciones que plantea que se identificaron valores de frecuencia mayores (72,2 %). 17
Los resultados del presente trabajo se corresponden con lo descrito por otros autores revisados en que la frecuencia del origen desconocido, es siempre mayor en estudios postnatales. Aunque, la cifra obtenida fue mayor (82,5 %) a lo reportado por otros autores, que publican números entre 40 % y 50 %.14,20 Se considera esté relacionado con el hecho de que los pacientes adultos se estudiaron solo a ellos, y no se extendió el estudio citogenético a los padres u a otros miembros de la familia, por lo que no se pudo identificar el origen de los mismos.
En los estudios prenatales la cifra de frecuencia (17,5 %) aumentó ligero, esta no se incluye en los valores registrados en los artículos revisados que muestran un rango entre 5 % y 17 %.13,21,22 Esto es debido a que, cuando se detecta una alteración estructural en estos estudios, siempre se indica el análisis cromosómico a los padres para conocer el origen de la anomalía y de esta manera realizar un asesoramiento genético correcto a la pareja. La cifra leve, aumentada en esta muestra, está basada en que, uno de los padres no se presentó para realizarse el estudio cromosómico indicado.
La incidencia de los reordenamientos estructurales prenatales “de novo” (1,1/1000) fue 0,4 veces menor a la reportada por Hook EB , en su estudio que determinó una frecuencia de 2,9/1000.13
Se concluye que, el origen de los reordenamientos estructurales de causa desconocido y en estudios postnatales predominó. Se identificaron en estudios prenatales las mayores frecuencias de los orígenes heredados y de “de novo” que evidencia que por las características de estos estudios hubo un seguimiento más directo y efectivo en cuanto a identificar el origen del RCE. Incrementar el conocimiento y la detección del origen de los portadores de RCE mediante la extensión del estudio a los padres en primer lugar, y a otros miembros de la familia de portadores, serviría para poder brindar y establecer estrategias de prevenciones directas y específicas en cuanto a la planificación familiar y asesoramiento genético específico y personalizado. Esto indica que aún quedan aspectos a mejorar para brindar cada día servicios de genética óptimos.
Agradecimientos
A todos los trabajadores de los laboratorios de Citogenética de todas las etapas, de los Centros Nacional de Genética y Provincial de Genética de La Habana, que con gran desempeño y dedicación contribuyeron con los diagnósticos cromosómicos. A los especialistas de Genética Clínica y Másteres en Asesoramiento Genético en La Habana.
REFERENCIAS BIBLIOGRÁFICAS
1. Iain D O'Neill. Homozygosity for constitutional chromosomal rearrangements: a systematic review with reference to origin, ascertainment and phenotype. Journal of Human Genetics. 2010;55:559-64.
2. Charleen M, Robert B. Chromosomal Genetic Disease: Structural Aberrations. ENCYCLOPEDIA OF LIFE SCIENCES/Nature Publishing Group; 2001. p. 1-8. [Citado: 17 de julio de 2014]. Disponible en: http: //www.els.net
3. Emery and Rimon’s. Principles and Practice of Medical Genetics. Fourth edition: Churchill Livingstone; 2002.
4. Thompson MW, Mclness RR, Wiliard HF. Citogenética clásica: principios generales y anomalías autosómicas. En: Masson editor. Genética en Medicina. 4a ed. Barcelona: Masson. 1996:191-218.
5. Recasens MM, Estival Calleja X. Anomalías cromosómicas. En Medicina Interna; 1995. p. 1214-22.
6. Heredero L. Comprehensive national genetic program in a developing country in Cuba: Kuliev A, Greendale K, Penchaszadeh VB, Paul NW (Eds): Genetics Services Provision: An International Perspective. Birth Defects Origin Art Ser. 1992;28(3):52-7.
7. Menéndez F, Casaña H, Quintana J, Quiñones O, Méndez LA, Nazabal I, et al. Cytogenetic Prenatal Diagnosis in Havana, Cuba. Am J Hum Genet.1987(Suplement);41:3 .
8. Quintana J, Quiñones O, Méndez L, Lavista G, Gómez M, Dieppa N, et al. Resultados del diagnóstico Prenatal Citogenético en las provincias occidentales de Cuba, 1984-1998. Rev Cubana Gen Hum.1999;1(3):2-8.
9. Méndez Rosado LA, Hernández Pérez G, Palencia Céspedes D, Quiñones Maza O, Barrios Martínez A, Suárez Mayedo U, et al. Mosaicismo de aberraciones estructurales, incidencia y repercusión prenatal. Rev Cub Gen Comunit. 2007;1(1):34-6.
10. Méndez L, Lantigua A, Quiñones O, Barrios A, Soriano M, González N, et al. Aplicación de una estrategia en el diagnóstico prenatal del mosaicismo cromosómico para la eliminación de falsos positivos. Rev Cubana Gen Comunit. 2011;5(1).
11. Marcheco BT. Genética Comunitaria: la principal prioridad para la genética médica en Cuba. Rev Cubana Gen Comun.2008;2(3):1.
12. Quiñones Maza O, Quintana Aguilar J, Méndez Rosado LA, Barrios Mesa A, Suárez Mayedo U, García Rodríguez M, et al. Frecuencias de reordenamientos cromosómicos estructurales acorde a las indicaciones para estudios citogenéticos prenatales y postnatales. Rev Cubana Genet Comunit.2010;4(3):36-42.
13. Hook EB, Schreinemachers DM, Wiiley AM, Cross PK. Inherited structural cytogenetic abnormalities detected incidentally in fetus diagnosed prenatally: frecuency, parental age associations, sex-ratio trends, and comparison with rates of mutants. Am J Hum Genet. 1984 Mar;36(2):422-43.
14. Prieto Carrasquero M, Rojas Atencio A, González S, Pineda Del Villar L, Soto M, Quintero M, et al. Clinical experience with balanced reciprocal translocation. Genet Couns. 1995;6(4):349-54.
15. Youings S, Ellis K, Ennis S, Barber J, Jacobs P. A study of reciprocal translocations and inversions detected by light microscopy with special reference to origin, segregation and recurrent abnormalities. Am J Med Genet. 2004 Abr;126(1):46-60.
16. Forrester MB, Merz RD. Patterns of chromosomal inversions identified by a birth defects registry, Hawaii, 1986-2002. Congenit Anom (Kyoto). 2007 Sep;47(3):97-100.
17. Forrester MB, Merz RD. Patterns of chromosomal deletions identified by a birth defects registry, Hawaii, 1986-2003. Congenit Anom (Kyoto). 2007 Jun;47(2):58-62.
18. Moreno García, FJ Fernández Martínez, E Barreiro Miranda. Repercusión clínica de las anomalías cromosómicas. An Pediat (Barc). 2004;61:236-41.
19. Peng HH, Chao AS, Wang TH, Chang SD. Prenatally diagnosed balanced chromosome rearrangements: eight years experience. J Reprod Med. 2006 Sep;51(9):699-703.
20. Forrester MB, Merz RD. Patterns of chromosomal translocations identified by a birth defects registry, Hawaii, 1986-2000. Genet Test. 2004 Summer;8(2):204-8.
21. Caron L, Tihy F, Dallaire L. Frequencies of chromosomal abnormalities at amniocentesis: over 20 years of cytogenetic analyses in one laboratory. Am J Med Genet. 1999 Jan;82(2):149-54.
Recibido: 1 de agosto de 2014.
Aprobado: 3 de octubre de 2014.
Olga Luisa Quiñones Maza . Centro Provincial de Genética Médica. La Habana, Cuba. Correo electrónico: olgaq@infomed.sld.cu


{kind=link}
{kind=link}
{kind=link}