










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter v.23 n.1 Ciudad de la Habana ene.-abr. 2007
Presentación de casos
Instituto de Hematología e Inmunología
Aplasia irreversible por el tratamiento con mesilato de Imatinib en una leucemia mieloide crónica. Presentación de un caso
Dra. Olga Agramonte Llanes, Dra. Valia Pavón Morán, Dr. Carlos Hernández Padrón, Dr. Rafael Losada Buchillón, Dr. José Mesa Cuervo, Dr. Luis Gabriel Ramón Rodríguez, Dr. Onel Ávila Cabrera y Dr. Roberto Silva Aguiar
Resumen
Se presenta una paciente de 45 años de edad diagnosticada en marzo de 1984 como una leucemia mieloide crónica Ph + , BCR/ABL positivo, que llevó tratamiento con busulfán, hidroxiurea, interferón y arabinósido de citosina durante 15 años. En marzo del 2003 se diagnosticó fase de transformación y en abril se comenzó la administración de Imatinib en dosis de 600mg diarios. Evolutivamente presentó dolores óseos ligeros, edema palpebral y en el día 35 pancitopenia severa, que provocó la suspensión del tratamiento. Se tomaron muestras para medulograma y biopsia de médula ósea y se diagnosticó una aplasia medular severa. Se administró tratamiento con antibioticoterapia de amplio espectro, hemoderivados y factor estimulador de colonias granulocíticas. A pesar de estas medidas terapéuticas, la paciente falleció a los 46 días de suspendido el tratamiento con Imatinib, con un cuadro clínico de aplasia medular irreversible y distrés respiratorio, complicaciones atribuibles al Imatinib.
Palabras clave: aplasia irreversible, mesilato de imatinib, leucemia mieloide crónica.
La leucemia mieloide crónica (LMC) es un desorden clonal del stem cell hematopoyético caracterizado por una translocación cromosomal recíproca que involucra a los cromosomas 9 y 22, t (9,22) que origina al cromosoma de Filadelfia (Ph) y fusiona a los genes bcr/abl. Esto da lugar a una expresión constitutiva de una proteína quimérica, bcr-abl, con aumentada actividad tirosinaquinasa.1 Los efectos que provoca esta activación puede incluir una proliferación independiente de factores de crecimiento, defecto en la adhesión celular y disminución de la apoptosis de las células hematopoyéticas.2-4 El mesilato de Imatinib (MI) es un inhibidor selectivo de la actividad tirosinaquinasa del BCR-ABL pues bloquea el sitio de unión para el ATP de la quinasa ABL, por lo tanto, impide la fosforilación de la tirosina en la proteína sustrato.5,6 Recientes reportes han demostrado la eficacia del mesilato de Imatinib (MI) en el tratamiento de la LMC, tanto de la fase crónica (FC) como en la fase acelerada (FA) y en la crisis blástica (CB).7,8 El MI induce marcados cambios morfológicos en la médula ósea, y en un gran número de pacientes la médula adquiere una apariencia indistinguible de la médula ósea normal. Otros pacientes exhiben una relativa hiperplasia eritroide no usual en la LMC.9 Además, reduce la celularidad de la médula ósea en todas las fases de la LMC y puede provocar, en algunos casos, una hipoplasia severa con mayor frecuencia en las fases avanzadas de la enfermedad. Por otra parte, disminuye la fibrosis reticulínica de la médula ósea.10
Los cambios de la médula ósea pueden ocurrir independientemente de la respuesta citogenética, lo que sugiere que estos hallazgos no se deben solamente a la erradicación de la hematopoyesis Ph+ y la reconstitución de la médula ósea normal.11
La base para la respuesta clínica al MI se presume sea la inhibición del crecimiento y la apoptosis de las células de la LMC que expresan la proteína quimérica bcr/abl.12,13 Además, esta inhibición puede afectar la diferenciación y el funcionamiento de las células leucémicas.14,15
La restauración de la hematopoyesis normal durante el tratamiento con MI depende en parte de la capacidad de los stem cell normales residuales para repoblar la médula ósea.14
En este trabajo comunicamos los datos más sobresalientes observados en una paciente con LMC, que presentó una aplasia medular irreversible durante el tratamiento con MI.
Presentación del caso
Paciente femenina, de 45 años, mestiza, que en marzo de 1984 se le diagnosticó una leucemia mieloide crónica (LMC) y que solo aquejaba astenia marcada. En el examen físico se encontró palidez cutáneo-mucosa y hepatoesplenomegalia de 3cm. Las pruebas de laboratorio mostraron hemograma: Hb 106g/L, reticulocitos 2,4 %, plaquetas 320x109 /L, leucocitos 60x109 /L, blastos 4 %, mielocitos 6b %, juveniles 5 %, stabs 2 %, segmentados 69 %, linfocitos 7 %, eosinófilos 3 %, monocitos 3 %, basófilos 1 %. El medulograma inicial y la biopsia de medula ósea concluyeron como una LMC en fase crónica (fig. 1).
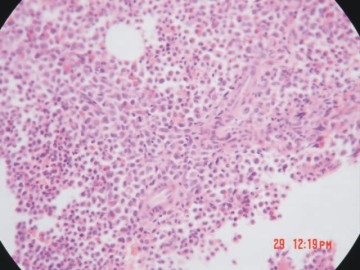
Fig. 1. Biopsia de medula ósea de una leucemia mieloide crónica en fase crónica.
El estudio citogenético evidenció el cromosoma Ph
Se inició tratamiento con busulfán, y una vez alcanzada la remisión hematológica se inició la terapia de mantenimiento con Interferón alfa recombinante (Heberon Alfa R, Heber Biotec) en dosis de 3 millones de unidades / m2 de superficie corporal, 3 veces por semana.
En octubre del 2001 presentó leucocitosis ligera y se decidió aumentar la dosis de INF a R a 6 millones de unidades/m2, 3 veces por semana, además de arabinósido de citosina (ARA-C) en dosis de 100mg/diarios por 5 días en forma de ciclos mensuales. Después de 4 ciclos de INF a R + ARA-C, se logró normalizar el hemograma, que se mantuvo dentro de los parámetros normales durante 3 meses. Las manifestaciones adversas a la terapia con ARA-C fueron severas, por lo que se decidió la suspensión de este medicamento.
En enero del 2003 presentó leucocitosis de 15x109 /L, con presencia del 10 % de mielocitos y al examen físico, se detectó esplenomegalia. Se realizó medulograma, que fue compatible con el diagnóstico de LMC en fase de transformación. La biopsia de médula ósea mostró una celularidad del 98-99 %, con marcados cambios hiperplásticos del sistema granulopoyético, y en menor intensidad del sistema megacariopoyético. Se observó además, depresión eritropoyética, incremento ligero del retículo y presencia de nidos de mieloblastos. Se concluyó como una LMC en transformación (fig. 2).
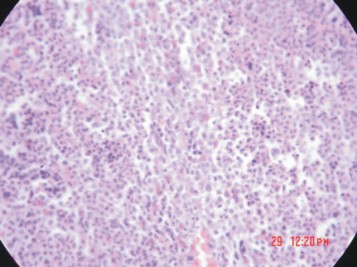
Fig. 2. Biopsia de medula ósea de una LMC en fase acelerada.

Fig. 3. Biopsia de medula ósea de una LMC en aplasia.
Se suspendió la administración del INF a R y el 24 de marzo del 2004 se comenzó tratamiento con MI (Glivec, Novartis), en dosis de 600mg diarios. La visceromegalia desapareció y solo presentó ligeras reacciones secundarias al medicamento, expresadas en dolores musculares y artralgias. Se mantuvo con el hemograma normal y el estado general aceptable durante 34 días, pero al día 35 del tratamiento manifestó toma ligera del estado general y lesiones purpúrico-hemorrágicas diseminadas por todo el cuerpo. El hemograma mostró: Hb 117g/L, plaquetas 18x109 /L y leucocitos 3,6x109 /L, segmentados 69 %, linfocitos 29 % y eosinófilos 2 %. Se suspendió la administración del MI en espera de la recuperación de la trombocitopenia severa y se hizo un medulograma en el que se obtuvo escaso material, celularidad disminuida, depresión de los sistemas megacariopoyético y granulopoyético e hiperplasia del sistema eritropoyético, con células blásticas <5 %. En la biopsia de médula ósea se apreció celularidad del 30-40 % con marcada depresión de los sistemas granulopoyético y megacariopoyético, compatible con una hipoplasia medular secundaria al tratamiento con MI. Clínicamente, la paciente se mantuvo sin variaciones en su cuadro clínico, pero con trombocitopenia y leucopenia severa, por lo que se ingresó y se administraron concentrados de plaquetas, antibioticoterapia de amplio espectro profiláctica y factor estimulador de colonias granulocíticas (FEC) (LeukoCim, CIMAB SA, La Habana). Evolutivamente mostró toma del estado general, sangramiento localizado en la mucosa oral, taquicardia y edemas en miembros inferiores.
Posteriormente comenzó con dolor en punta de costado en la base de hemitórax derecho y disnea ligera.
En el estudio radiográfico de tórax se observó un discreto infiltrado inflamatorio en ambos campos pulmonares, por lo que se inició la administración de ceftriaxona y amikacina por vía endovenosa, además de inmunoglobulina G (Intacglobin) (Empresa Productora de Sueros y Productos Hemoderivados Adalberto Pessant González) en dosis de 400mg/kg de peso durante 5 días, y se mantuvo la terapéutica con transfusiones de plaquetas, glóbulos y FEC-G. En hemogramas evolutivos se observó disminución progresiva del conteo de leucocitos y la neutropenia llegó a valores por debajo de 0,3x109 /L. A los 37 días de suspendido el MI se repitió el medulograma y la biopsia de médula ósea, los que confirmó el diagnóstico de aplasia medular (fig. 3). En el hemograma realizado un día después se encontró: Hb 82g/L, leucocitos 0,05x109 /L, plaquetas 2 x109 /L; la función hepática y renal se mantuvieron estables. La paciente se mantuvo afebril y las manifestaciones purpúricas en la piel aumentaron. El cuadro respiratorio empeoró a pesar del tratamiento con manifestaciones clínicas de distrés respiratorio, que motivó la intubación endotraqueal con respiración asistida. La paciente fallece a los 72 días de iniciado el tratamiento con MI, en un cuadro de aplasia irreversible atribuible a este medicamento y distrés respiratorio.
El estudio necrópsico mostró aplasia medular severa, pancreatitis aguda comenzante, necrosis tubular aguda, plasmocitosis renal ligera, hemorragias endocárdicas y miocárdicas extensas, pleuritis aguda reactiva, hemorragias subaracnoideas multifocales de convexidad y hemorragias focales intraparenquimatosas bilaterales.
Discusión
La LMC es, probablemente, la enfermedad maligna más ampliamente estudiada.16-18 Antes del advenimiento del MI, la vida media de los enfermos era de 5-6 años después del diagnóstico. Sin embargo, algunos tienen un curso mucho más agresivo desde el comienzo de la enfermedad y mueren en el primer año del diagnóstico, mientras que otros sobreviven por 20 años o más. Esto presupone que eventos genéticos secundarios pueden suplantar el efecto del cromosoma Ph.19
El tratamiento de primera línea para pacientes con LMC previo a la introducción del MI, incluía la hidroxiurea y regímenes basados en la terapia con INF a R, solo o combinado con ARA-C, pero ninguno reconocido como curativo.20
El trasplante de médula ósea alogénico y de células progenitoras hematopoyéticas obtenidas de la sangre periférica, es considerado aún la única terapia curativa para esta enfermedad, pero solamente una minoría de los pacientes son elegibles para el mismo, además de que los riesgos de morbilidad y mortalidad directamente atribuible a este proceder resultan apreciables.21
Las opciones de tratamiento para pacientes con diagnóstico reciente han cambiado desde la introducción del MI. Hasta hace muy poco tiempo, el INF fue aceptado como el mejor agente terapéutico para tratar pacientes con LMC en fase crónica no elegibles para trasplante de médula ósea alogénico. El INF induce respuesta citogenética completa o parcial en un 10-30 % de los pacientes, y probablemente, prolonga la supervivencia mucho más que la hidroxiurea, pero puede causar un amplio espectro de efectos secundarios, especialmente en ancianos.22-28 Estudios multicéntricos han mostrado que la sobrevida de los pacientes tratados con la combinación de INF y Ara-C es superior a la de los pacientes tratados solo con INF.29,30
El MI es una pequeña molécula inhibidora de las señales de transducción; su acción específica va dirigida a una serie de proteinaquinasas (ABL, c-KIT), receptor del factor de crecimiento derivado de plaquetas (PDGR-R) y sus formas oncogénicas, más notablemente el bcr-abl. 31-35 El MI representa el arquetipo de una nueva clase de agentes anticancerígenos, que no son más que pequeñas moléculas con una alta selectividad dirigidas a dianas moleculares específicas responsables del establecimiento y mantenimiento del fenotipo maligno.36
En la LMC la eficacia del MI no tiene precedentes, con índices de respuesta hematológica completa (RHC) aproximadamente en el 100 % de los pacientes en fase crónica.37
A pesar de que la terapia con MI es bien tolerada, el uso de este medicamento no está libre de efectos secundarios, particularmente náuseas, edema, fatiga, cefalea, calambres musculares, artralgias, mialgias, diarreas, erupción cutáneo y mielosupresión.35-37 La mielosupresión es uno de los efectos secundarios más severos en los pacientes con LMC y es común en pacientes con fases avanzadas de la enfermedad, ya sea en FA o CB. Usualmente se inicia dentro de las semanas 2-4 de comenzada la terapia en la CB y ligeramente más tarde en la FA.37 Se ha señalado que la CB induce la mielosupresión en el 1 % de los pacientes 38,39 y puede ocurrir en cualquier momento durante el tratamiento.
En nuestra paciente, el inicio de la mielosupresión se corresponde con lo comunicado por otros autores. Se han señalado algunas características clínicas asociadas con un mayor riesgo de mielosupresión que incluyen un alto porcentaje de blastos en la médula ósea, bajos niveles de hemoglobina y citopenias inducidas por el INF.40,41 Estas características no las presentó nuestro caso; sin embargo, sí evidenció otros factores de riesgo como es el antecedente de un largo período de evolución y terapia previa con busulfán, además de un prolongado tratamiento con INF.
Se ha reportado que una minoría de pacientes (<5 %), experimenta episodios repetidos de mielosupresión, por lo que en ellos se requiere suprimir el tratamiento por un tiempo prolongado, lo que se asocia con un alto riesgo de recaídas.42 En nuestro caso, la terapia solo fue suspendida al presentar trombocitopenia muy severa y neutropenia severa. A pesar del tratamiento de apoyo con FEC-G, no se logró la recuperación de la paciente y falleció en un cuadro clínico y hematológico de aplasia medular a los 46 días de suspendido el tratamiento.
El uso de los factores estimuladores de colonias en los enfermos con fases avanzadas de la enfermedad durante la mielosupresión, todavía resulta controvertido, pero se justifica en casos como este, en el que la citopenia es por aplasia medular y no por progresión de la enfermedad. De hecho, estos pacientes no experimentan altos índices de recaídas, lo que sugiere que los factores estimuladores del crecimiento mieloide no afectan adversamente la actividad antileucémica del MI.43,44
En pacientes con LMC, la mayor parte de la hematopoyesis deriva del stem cell Ph+, y a medida que progresa la enfermedad, el compartimiento de células progenitoras gradualmente resulta dominado por las células Ph positivas.45 Tanto in vivo como in vitro, existen datos que indican que el MI no afecta severamente la hematopoyesis residual normal y, por lo tanto, las dosis terapéuticas del medicamento inhiben tan solo del 10-20 % la formación de colonias por células progenitoras normales.46-49
Se conoce que los stem cell de la LMC paradójicamente muestran una menor habilidad para implantarse comparados con las células normales, lo que sugiere que por inhibición de la proliferación de las células leucémicas, el MI puede permitir el desarrollo de una hematopoyesis normal Ph- sobre la hematopoyesis Ph+.30,31 Sin embargo, Hasserjian y colaboradores 11 observaron varios casos en los cuales las células Ph- no lograron emerger en una médula ósea normocelular o con una hipoplasia compensada secundaria al uso del MI. Esto refleja deficiencia en los stem cells normales o un daño estromal de la médula ósea, debido a una intensa terapia o a una prolongada estancia en la médula de las células de la LMC, lo que sugiere que el MI puede ser más efectivo cuando se utiliza tempranamente en la fase crónica de la LMC.26
En nuestra paciente, por el tiempo de evolución de la enfermedad y encontrarse en fase acelerada, consideramos que el grado de hematopoyesis residual normal sería mínimo o prácticamente nulo, lo que añadido al efecto mielosupresor del MI, nos podría justificar el tiempo de aplasia posmedicamentosa y la no recuperación hematológica, a pesar del tratamiento de apoyo recibido. Este es el reporte del primer caso en Cuba de aplasia medular irreversible secundario al uso del MI. Hasta donde conocemos, esta es una complicación poco frecuente, pero que debe mantenerse en mente como un posible efecto secundario del MI.
Summary
Irreversible aplasia due to the treatment with imatinib mesilate in a chronic myeloid leukemia. A case report
A 45-year-old female patient who was diagnosed chronic myeloid leukemia Ph+ in March 1984, and had treatment with busulfan, hydroxyurea, interferon and cytosine arabinoside during 15 years is presented. In March 2003, the transformation stage was diagnosed and, in April, she began to receive imatinib at daily doses of 600 mg. Evolutively, she had mild bone pain, palpebral edema and, on the 35th day, severe pancytopenia that caused the suspension of the treatment. Bone marrow samples were taken by aspiration and biopsy, and a severe medular aplasia was diagnosed. Treatment with wide-spectrum antibiotic therapy, hemoderivates, and granulocyte colony-stimulating factor was applied. In spite of these therapeutic measures, the patient died 46 days after interrupting the treatment with imatinib, with a clinical picture of irreversible medular aplasia and respiratory distress, complications attributable to Imatinib.
Key words: irreversible aplasia, imatinib mesylate, chronic myeloid leukemia.
Referencias bibliográficas
1. Deininger WM, Vieira S, Mendiola R, Schultheis B, Goldman JM, Melo JV. BCR-ABL tyrosine kinase activity regulates the expression of multiple genes implicated in the pathogenesis of chronic myeloid leukemia. Cancer Res 200;60:2049-55.
2. Marley SB, Deininger MWN, Davidson RJ, Goldman RJ, Gordon MY. The tyrosine kinase inhibitor STI571, like interferon alpha, preferentially reduces the capacity for amplification of granulocyte –microphage progenitors from patients with chronic myeloid leukemia. Exp Haematol 2000;28:551-7.
3. Oetzel C, Jonuleit T, Gotz A, Kuip H, Michels H, Duyster J, et al. The tyrosine kinase inhibitor CGP 57148(STI 571) induces apoptosis in BCR-ABL positive cells by down regulating BCL-X:Clin Cancer Res 2000;6:1958-68.
4. Gaston I, Stenberg PE, Bhat A, Druker BJ. Abl kinase by not P13- kinase links to the cytoeskeletal defects in Bcr-Abl transformed cells. Exp Haematol 2000;28:77-86.
5. Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kurivan J. Structural mechanism for STI 571 inhibition of Abelson tyrosine kinase. Science 2000;289:1938-42.
6. Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, et al. Effects of a selective inhibition of the Abl tyrosine kinase on the growth of BCR-ABL positive cells. Nat Med 1996;2:561-6.
7. Druker BJ, Talpaz M, Resta D, Peng B, Buchdunger E, Ford JM, et al. Efficacy and safety of a specific inhibitor of the BCR/ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 2001;344:1031-7.
8. Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Fernandez S, Ford JM, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med 2001;344:1038-42.
9. Facchetti F, Tironi A, Marocolo D. Histopathological changes in bone marrow biopsies from patients with chronic myeloid leukemia after treatment with recombinant alpha interferon. Histopathology 1997;31:3-11.
10. Loran –Metze I, Vassallo J, Souza CA. Histological and cytological heterogeneity of bone marrow in Philadelphia –positive chronic myelogenous leukaemia at diagnosis. Br J Haematol 1987;67:45-9.
11. Hasserjian RP, Boecklin F, Parker S, Chase A, Dhar S, Zaiac M, et al. STI571(Imatinib Mesylate) Reduces Bone Marrow Cellularity and Normalizes Morphologic Features Irrespective of Cytogenetic Response. Am J Clin Pathol 2003;113:142-8.
12. Gambacorti-PasseriniC, Le Coutre P, Mologni L, Marchesi E. Inhibition of de ABL kinase activity blocks the proliferation of BCR/ABL positive leucemic and induces apoptosis. Blood Cells Mol Dis 1997;23:380-94.
13. Deininger WM, Goldman JM, Lydon N, Vieira S, Mendiola R, Schultheis B. The tyrosine kinase inhibitor CGP57148B selectively inhibits the grow of BCR –ABL positive cells. Blood 1997;90:3691-8.
14. Bellucci R, Sala R, De Propis MS. Interferon Alpha and bcr-abl antisense oligonucleotides in combination enhance the antilekemia effect and the adherence of CML progenitors to performed stroma. Leuk Lymphoma.1999;35:471-81.
15. Dazzi F, Hasserjian R, Gordon MY, Apperley JF, Chase A, Lampert I. Normal and chronic phase CML hematopoietic cells repopulate NOD/SCID bone marrow with different kinetics and cell lineage representation. J Hematol 2001;1:303-15.
16. Michael W, Deninger N, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood 2000,96:3343-56.
17. Kantarjian HM, Deisseroth A, Kurzrock R, Estrov Z, Talpaz M. Chronic myelogenous leukemia: a concise update .Blood 1993;82:691-703.
18. Sokal JE, Baccarini M, Russo D, Tura S. Staging and prognosis in chronic myelogenous leukemia . Semin Hematol 1998;25:49-61.
19. Kurzrock R, Hagop M, Kantarjian M, Druker B, Talpaz M. Philadelphia chromosome-positive leukemias: From basic mechanisms to molecular therapeutics. Ann Intern Med 2003;138:819-30.
20. Bonifazi F, Vitro D, Rosti G, Guilhot F, Guilhot J, Trabacche E . Chronic myeloid leukaemia and interferon alfa: A study of complete cytogenetic responders. Blood 2001;98:3074-81.
21. Silver RT, Woolf SH, Hehlmann R, Appelbaum FR, Anderson J, Bennett C. An evidence -based analysis of the effect of busulfan, Hydroxyurea, interferon and allogeneic bone marrow transplantation in treating the chronic phase of chronic myeloid leukemia: Developed for American Society of Hematology . Blood 1999;94:1517-36.
22. Chronic Myeloid Leukemia Trialists Collaborative Group. Interferon alfa versus chemotherapy for chronic myeloid leukemia: a meta- analysis of seven randomized trials. J Natl Cancer Inst 1997;89:1616-20
23. Sawyers CL. Chronic myeloid leukemia. N Engl J Med 1999;340:1330-40.
24. Goldman JM, Druker BJ. Chronic myeloid leukemia: Current treatment options. Blood 2001; 98:2030-42.
25. Borden EC, Parkinson D. A perspective on the clinical effectiveness and tolerance of interferon-alfa. Semin Oncol 1998; 25:39-47.
26. Trask P, Esper P, Riba M, Redman B. Psychiatric side effects of interferon therapy: Prevalence, proposed mechanism, and future directions. J Clinic Oncol 2000;18:2316-26.
27. Motzer RJ, Murphy BA, Bacik J, Schwartz LH, Nanus DM, Mariani T. Phase III trial of interferon 2ª with or without 13 cis –retinoic acid for patients with advanced renal cell carcinoma. J Clin Oncol 2000;18:2972-80.
28. Bacik J, Mazundar M, Fairclough DL, Eremenco S, Mariani T, Motzer RJ. The functional assessment of cancer therapy-BRM(FACT-BRM). A new tool for the assessment of quality of life treated with biologic responser modifiers. Qual Life Res 2004;13:137-54.
29. Guilhot F, Chastang C, Michallet M, Guerci A, Harousseau JL, Maloisel F. Interferon alfa 2b combined with cytarabine versus interferon alone in chronic myelogenous leukemia. N Engl J Med 1997;337:223-9.
30. Baccarini M, Rosti G, de Vivo A , Bonifazi F, Russo D, Martinelli G. A randomized study of interferon alfa versus interferon alfa and low dose arabinosyl cytosine in chronic myeloid leukemia. Blood 2002;99:1527-35.
31. Buchdunger E, Zimmermann J, Mett H, Meyer T, Muller M, Regenass U . Selective inhibition of the platelet-derived growth factor signal transduction pathway by a protein –tyrosine kinase inhibitor of the 2-phenylaminopyrimidine class. Proc Natl Acad Sci USA 1995;92:2558-62.
32. Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S l. Effects of selective inhibition of the ABL tyrosin kinase on the growth of BCR-ABL positive cells. Nature Med 1996;2:561-6.
33. Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA, Zigler A J . Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood 2000;96:925-32.
34. Okuda K, Weisberg E, Gililand DG, Griffin JD. ARG tyrosine kinase activity inhibited by STI 571. Blood 2001;97:2440-8.
35. Beran M, Cao X, Estrov Z, Jeha S, Jin G, O'Brien E . Selective inhibition of cell proliferation and bcr-abl phosphorylation in acute lymphoblastic leukemia cells expressing Mr 1980, 000 BCR-ABL protein by a tyrosin kinase inhibnitor (CGP-57148). Clin Cancer Res 1998;4:1661-72.
36. Deininger MW, O ' Brien SG, Ford JM, Druker BJ . Practical management of patients with chronic myeloid leukemia receiving Imatinib. J Clin Oncol 2003;8:1637-47.
37. Kantarjian H, Sawyers CL, Hochhaus A , Guilhot F, Schiffer C, Gambacorti-Passerini C. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med 2002;346:645-52.
38. Sawyers CL, Hochhaus A, Feldman E, Goldman JM, Miller CB, Ottmann OG . Imatinib induces hematologic and citogenetic responses in patients with chronic myeloid leukemia in myeloid blast crisis. Results of phase II study. Blood 2002;99:3530-9.
39. Talpaz M, Silver RT, Druker BJ, Goldman JM, Gambacorti-Passerini C, Guilhot F, et al. Imatinib induces durable hematologic and cytogenetic responses in patients with accelerated phase chronic myeloid leukemia: Results of a phase 2 study. Blood 2002;99:1928-37.
40. Mauro MJ, Michael E, O'Dwyer E, Kurilik G, Blasdel C, Farnsworth M. Risk factors for mielosupresion in chronic phase CML patients treated with imatinib mesylate(STI): Blood 98:139 a, 2001(abstr 585).
41. Marktel S, Marin D, Foot N, Szydlo R, Bua M, Karadimitris A, et al. Chronic myeloid leukemia in chronic phase responding to imatinib: The occurrence of additional cytogenetics abnormalities predicts disease progression. Haematologica 2003;88:260-2.
42. Deininger MW, Goldman JM, Lydon N, Melo JV. The tyrosine kinase inhibitor CGP 57148B selectively inhibits the grow of BCR-ABL positive cells. Blood 1997;90:3691-8.
43. Mauro MJ , Kurilik G, Balleisen S, ODwyer ME ,Fernandes-Reese S, Druker BJ. Myeloid growth factors for neutropenia during imatinib mesylate (STI 571) therapy for CML: Preliminary evidence of safety and efficacy. Blood 2001;98:139a(Abstract).
44. Marin D, Marktel S, Foot N, Bua M, Goldman JM, Apperley JF.GCSF reverses cytopenias and permit cytogenetic responses in patients with chronic myeloid leukemia treated with imatinib mesylate. Haematologica 2003;88:227-9.
45. Petzer AL, Eaves CJ, Lansdorp PM. Characterization of primitive subpopulations of normal and leukemic cells present in the blood of patients with newly diagnosed as well as established chronic myeloid leukemia. Blood 1996;88:2162-71.
46. Van Oosterom AT, Judson I, Verweij J, Stroobants S, Donato di Paola E, Dimitrievic S. S afety and efficacy of imatinib (STI571) in metastatic gastrointestinal tumors: A phase I study. Lancet 2001;358:1421-3.
47. Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ. Efficacy and safety of the imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002 347:472-80.
48. Chng WJ, Tan LH. Late-onset marrow aplasia due to imatinib in newly diagnosed chronic phase chronic myeloid leukaemia. Leuk Res 2005;29:719-20.
49. Lemarbre G, Schinstock C, Hoyer R, Krook J, Tefferi A. Late onset aplastic anemia during treatment of chronic myeloid leukemia with imatinib mesylate. Leuk Res 2006; (pre-publicación en online).
Recibido: 23 de noviembre del 2006. Aprobado: 12 de diciembre del 2006.
Dra. Olga Agramonte Llanes. Instituto de Hematología e Inmunología. Apartado Postal 8070, Ciudad de La Habana, CP 10800, Cuba. Tel (537) 6438268, 6438695, 6434214, Fax (537) 442334. e -mail: ihidir@hemato.sld.cu

