










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión impresa ISSN 0864-0289
Rev Cubana Hematol Inmunol Hemoter vol.30 no.1 Ciudad de la Habana ene.-mar. 2014
PRESENTACIÓN DE CASO
Síndrome de Evans Fisher asociado con esclerodermia
Fisher Evans syndrome associated with scleroderma
Dr. Adrián Romero-GonzálezI, Dra. Mildrey Gil-AgramonteII, Dr. Alfredo Ortiz-LabradaI, Dra. Madeleyne Tamayo-RodríguezI, Dr. Vladimir Felinciano-ÁlvarezI, Lic. Julio César Suárez-CastilloI, Dr. Julio H. Gutiérrez-LagoI
I Hospital Militar Central "Dr. Luis Díaz Soto", La Habana, Cuba.
II Hospital Militar Central "Dr. Carlos J. Finlay", La Habana, Cuba.
RESUMEN
El síndrome de Evans Fisher, descrito por primera vez en 1951, es un desorden autoinmune caracterizado por la presencia simultánea o secuencial de anemia hemolítica, trombocitopenia inmune y, en ocasiones, neutropenia inmune; con una prueba de antiglobulina directa positiva. Puede ser de causa primaria o secundaria a otras condiciones, como el lupus eritematoso sistémico, los síndromes linfoproliferativos o inmunodeficiencias primarias. Es muy rara su asociación con la esclerodermia. Con el término esclerodermia, que en sentido literal significa piel dura, se designa un grupo de enfermedades y síndromes que tienen como característica común la induración y el engrosamiento cutáneos. Se caracteriza por la presencia de un depósito excesivo de los componentes del tejido conjuntivo, expresado en forma de fibrosis hística, y por alteraciones estructurales del lecho vascular. Con un cuadro clínico muy amplio, afecta fundamentalmente la piel y ciertos órganos internos, como tubo digestivo, pulmón, corazón y riñón. Se presenta una paciente femenina de 75 años de edad, piel negra, con antecedentes de hipertensión arterial, diabetes mellitus tipo 2, cardiopatía isquémica y esclerodermia, esta última diagnosticada seis meses antes de su ingreso. Acudió por decaimiento marcado, palidez cutáneo-mucosa intensa y petequias generalizadas. En los estudios realizados se detectó anemia y trombocitopenia severas, reticulocitosis, prueba de antiglobulina directa positiva e hipercelularidad medular con hiperplasia severa de los sistemas megacariopoyético y eritropoyético. Se diagnosticó un síndrome de Evans Fisher y se trató con esteroides e inmunomoduladores; se logró la mejoría clínica y la remisión hematológica.
Palabras clave: Evans Fisher, anemia hemolítica, trombocitopenia, esclerodermia.
ABSTRACT
The Evans syndrome, first described in 1951, is an autoimmune disorder characterized by the simultaneous or sequential development of hemolytic anemia and immune thrombocytopenia or immune neutropenia. It may be of primary origin or secondary to other diseases or conditions such as systemic lupus erythematosus, lymphoproliferative disorders or primary immunodeficiencies. Its association with scleroderma is considered very rare. The word scleroderma, which literally means hard skin, designates a group of diseases and syndromes of common feature in induration and thickening the skin. It is characterized by the presence of excessive deposition of connective tissue components, expressed as histic fibrosis, and structural alterations of the vascular bed. With a broad clinical view, it primarily affects the skin and certain internal organs such as gastrointestinal tract, lung, heart and kidney. We present a 75 year-old female, black skin, with a history of hypertension, type 2 diabetes, ischemic heart disease and scleroderma, the latter diagnosed six months before admission. The patient referred marked weakness, pale skin and generalized petechiae. The complete blood count detected severe anemia, thrombocytopenia and reticulocytosis. Other studies showed positive direct Coombs test and severe hypercellularity. Evans Fisher syndrome was diagnosed and treated with steroids and immunomodulators; clinical improvement and hematologic remission was achieved.
Keywords: Evans Fisher, hemolytic anemia, thrombocytopenia, scleroderma.
INTRODUCCIÓN
El síndrome de Evans Fisher (SEF) es un trastorno infrecuente caracterizado por la asociación simultánea o secuencial de trombocitopenia inmune y anemia hemolítica autoinmune; en ocasiones, neutropenia inmune, con una prueba de antiglobulina directa positiva.1-3 Esta condición fue descrita por Robert Evans en 1951, quien reportó un espectro de combinaciones clínicas entre trombocitopenia inmune primaria y anemia hemolítica adquirida con etiologías similares de formación de autoanticuerpos.4 Se describe con más frecuencia en mujeres y tiene una mayor incidencia en adultos que en niños.5
Se ha reportado su asociación a otras enfermedades o trastornos como el lupus eritematoso sistémico, los síndromes linfoproliferativos y las inmunodeficiencias primarias. Debido a la baja incidencia de presentación con la esclerodermia se presenta este caso para contribuir a aumentar conocimiento general de la enfermedad.
PRESENTACIÓN DEL CASO
Paciente femenina de 75 años de edad, de piel negra, con antecedentes personales de hipertensión arterial, diabetes mellitus tipo 2, cardiopatía isquémica y esclerodermia, diagnosticada esta última seis meses antes de su ingreso.
En diciembre de 2011 acudió con un cuadro de decaimiento marcado, palidez cutáneo- mucosa intensa y petequias generalizadas, frialdad distal, anorexia y heces fecales oscuras. Se le realizó hemograma de urgencia donde se detectó una bicitopenia por lo que ingresó.
RESUTADOS DE LABORATORIO
Los exámenes realizados mostraron los siguientes resultados: hemoglobina 72 g/L (VN: 120-150 g/L); conteo de reticulocitos 23 x 10-3 (VN: 5-15 x 10-3); plaquetas 10 x 109/L (VN: 150-400 x 109 /L); leucocitos totales 10,4 x 109/L (VN: 4,5 - 11x 109 /L); neutrófilos 7,1 x 109 /L (VN: 1,8 7,5 x 109/L); linfocitos 2,7 x 109/L (VN: 1,0 4,8 x 109/L); eosinófilos 0,2 x 109 /L (VN: 0,04 0,4 x 109/L). Velocidad de sedimentación eritrocitaria: 23 mm/h (VN: hasta 20 mm/h).
Glicemia 7,45 mmol/L (VN: 3,2-6,2 mmol/L); creatinina 89 µmol/L (VN: 80-120 ìmol/L); uratos 446 µmol/L (VN: 140-440 ìmol/L); alaninoaminotransferasa 32 U/L (VN: hasta 40 U/L); aspartatoaminotransferasa 34 U/L (VN: hasta 40 U/L); deshidrogenasa láctica 980 U/L (VN: hasta 440 U/L); bilirrubina total 25,2 µmol/L (VN: 2-17 ìmol/L); bilirrubina directa 12 µmol/L (VN: hasta 7 ìmol/L); bilirrubina indirecta 13,2 µmol/L (VN: hasta 12 ìmol/L); proteínas totales 73,5 g/L (VN: 60-80 g/L).
Coagulograma: tiempo de sangramiento (método de Duke) 6 min (VN: 1 a 3 min); prueba del lazo positiva, coágulo irretráctil, tiempo de coagulación (método de Lee White) 8 min (VN: 5 a 10 min); tiempo de protrombina (método de Quick): control -13,2 s, paciente -14,8 s (VN: hasta 3 s por encima o por debajo del control); tiempo parcial de tromboplastina-Kaolin (método de Rappaport): control - 32,4 s; paciente - 34,6 s (VN: hasta 6 s por encima o por debajo del control).
Lámina periférica: anisocitosis, hipocromía, policromatofilia, microesferocitos, poiquilocitosis, punteado basófilo, anillos de Cabot, leucocitos adecuados, trombocitopenia severa con algunas macroplaquetas.
Prueba de antiglobulina directa: positiva
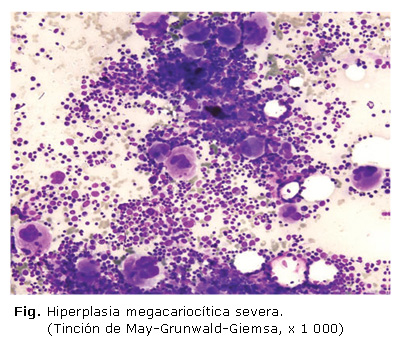
Medulograma: médula hipercelular con hiperplasia severa de los sistemas megacariopoyético y eritropoyético, este último con todas sus formas madurativas y con cambios megaloblásticos; se observaron abundantes nidos de normoblastos, no se constató infiltración medular; menos del 5 % de blastos de aspecto mieloide; Azul de Prusia positivo (figura).
Estudios de autoinmunidad: Determinación de anticuerpos (Acs) anti-ds ADN (IgG): 40 U/mL, VN (hasta 20 U/mL); Acs antitopoisomerasa -1: 87 U/mL, VN (hasta 25 U/mL); Acs anti ribonucleoproteína: 102 U/mL, VN (hasta 25 U/mL). Se determinó además: Acs ant - Smith; Acs anti - Ro; Acs anti - La; Acs anti - Jo; Acs anticardiolipina IgG/IgM, IgA; todos negativos. Factor reumatoideo: 12 UI/mL, VN (hasta 20 U/mL); proteína C reactiva: 46,8 mg/L VN (hasta 8 mg/L); complemento C3: 1,07 g/L, VN (0,75-1,35 g/L); complemento C4: 0,23 g/L, VN (0,09-0,36 g/L). Cuantificación de IgA, IgG, IgM dentro de parámetros normales.
Ecografía abdominal: hepatomegalia de 2 cm por debajo del reborde costal, de bordes finos bien definidos. Esplenomegalia de 183 x 91 mm (VN: hasta 140 x 70 mm). Resto de los órganos intrabdominales sin alteraciones. No se observaron adenopatías profundas.
Se concluyó que presentaba un síndrome de Evans Fisher asociado o secundario a la esclerodermia.
La paciente fue ingresada en el servicio de cuidados intermedios de medicina, donde recibió atención integral por un equipo médico multidisciplinario integrado por hematólogo, reumatólogo, clínicos e intensivistas.
Se inició tratamiento con gammaglobulina endovenosa (Intacglobin®, La Habana, Cuba) en dosis 1g/m2 por 2 días, posteriormente se transfundió con concentrado de plaquetas a razón de una unidad por cada 10 kg de peso cada 12 horas, y las manifestaciones hemorragíparas desaparecieron. Posteriormente, se comenzó tratamiento con metilprednisolona a 500 mg/m2 diarios durante 5 días. Se continuó con prednisona a 1 mg/kg/d; con lo que se logró la recuperación progresiva de las citopenias hasta su total normalización.
Se egresó y se mantiene en seguimiento por consulta externa con buen estado general, mejoría clínica y en remisión hematológica.
DISCUSIÓN
La esclerodermia sistémica (ES) es una enfermedad autoinmune crónica que afecta la piel; los órganos, especialmente pulmón y riñón, y el sistema musculoesquelético. La presencia de fenómeno de Raynaud, anticuerpos antinucleares positivos y capilaroscopía patológica son signos tempranos de la enfermedad. Los subtipos clínicos fundamentales son ES limitada, la ES difusa y los síndromes de superposición. Los pacientes con ES requieren un tratamiento multidisciplinario con un seguimiento estricto en aras de reconocer de forma temprana la afectación orgánica: neumopatía intersticial, hipertensión pulmonar, afectación renal, entre otras. El tratamiento es generalmente sintomático, pero deben utilizarse inmunosupresores cuando existe afectación orgánica.6
La esclerodermia se asocia muy raramente con el SEF; este último se observa más frecuentemente con otras enfermedades del tejido conectivo, particularmente con el lupus eritematoso sistémico,2,5
Robert Evans fue el primero en describir la asociación entre trombocitopenia inmune y anemia hemolítica autoinmune.3 Aunque su frecuencia no es exactamente conocida, se sabe que el SEF es un desorden extremadamente raro.7 En una revisión de pacientes adultos con inmunocitopenias entre 1950 y 1958, solo 6 de los 766 casos estudiados fueron diagnosticados con SEF.8 En la década de los años 60 del pasado siglo se estimó que la incidencia mínima anual en los Estados Unidos de América fue de 1:80 000 habitantes.3
La ocurrencia en pacientes de una misma familia se ha comunicado en muy escasos reportes.9 En el caso clínico que se presenta, no se recogieron antecedentes familiares. No existe predilección conocida, ya que se ha descrito en todos los grupos étnicos y en diferentes edades.10,11
Desde sus primeras descripciones se consideró y definió como un trastorno idiopático y su diagnóstico se ha basado en criterios de exclusión. Actualmente se ha podido determinar su asociación con numerosas enfermedades o condiciones como: conectivopatías, desórdenes linfoproliferativos e inmunodeficiencias primarias, o ambas.2,12-17
Las investigaciones han demostrado que el SEF es un trastorno de la regulación inmune generalizada, con la formación de autoanticuerpos dirigidos contra antígenos específicos de eritrocitos, plaquetas o neutrófilos, sin reacción cruzada.
Aunque nuestra paciente presentó cifras normales de inmunoglobulinas, está reportado que los pacientes muestran concentraciones disminuidas de inmunoglobulinas séricas (IgG, IgM, IgA), así como reducción de los linfocitos T auxiliadores e incremento de las células T supresoras. También se ha encontrado la producción aumentada de interleucina 10 y de interferón gamma, causantes de la producción de autoanticuerpos por las células B.1,8
Las manifestaciones clínicas están en relación con la anemia hemolítica: palidez cutáneo mucosa, ictericia, astenia e insuficiencia cardiaca en los casos severos y por otra parte, síntomas y signos secundarios a la trombocitopenia, como: petequias, hematomas, metrorragia, hematuria, sangramientos digestivos, en dependencia de la intensidad de la trombocitopenia. En nuestra paciente, las manifestaciones de bajo gasto fueron más evidentes dado no solo por la hemólisis sino por la hemorragia que contribuyó aún más a la intensificación de la anemia. Aunque la paciente presentó esplenomegalia gigante; se describe que los pacientes pueden presentar además adenopatías, hepatomegalia, o ambas.3,18
El diagnóstico se realiza por exclusión y debe ser bien definido para no llevar a un diagnóstico erróneo. Existen condiciones en las que esta entidad puede aparecer como causa secundaria; además de las ya mencionadas anteriormente podemos encontrar la enfermedad de Castleman, luego de la terapia recombinante con interleucina 2 en el carcinoma renal, o después de un trasplante de células hematopoyéticas, y en las que requiere un manejo diferente. Otras condiciones que causan anemia hemolítica y trombocitopenia deben ser diferenciadas de un SEF, entre las que se encuentran la hemoglobinuria paroxística nocturna, la púrpura trombocitopénica trombótica, la deficiencia de ADAMTS-13, el síndrome hemolítico urémico, el síndrome de Kasabach-Merrit, entre otros.8,19-22
Nuestra paciente tuvo una respuesta favorable con el uso de corticosteroides e inmunoglobulinas en las dosis habituales. Otras opciones de tratamiento para el SEF incluyen drogas inmunosupresoras como alcaloides de la vinca, danazol, así como esplenectomía y trasplante de células progenitoras hematopoyéticas.2,4,23 Recientemente, el uso del rituximab ha mostrado resultados promisorios.1,24,25
Hasta el momento la paciente se ha mantenido con buen estado general y en remisión hematológica, aunque continúa con la prueba de antiglobulina directa positiva.
Esta asociación de enfermedades es muy rara; hasta donde conocemos, no hay ningún reporte en Cuba, por lo que nuestra publicación es de gran interés para la comunidad médica al contribuir a ampliar y difundir los conocimientos sobre la asociación de estas dos enfermedades.
REFERENCIAS BIBLIOGRÁFICAS
1. Correa González LC. Desenmascarando el síndrome de Fisher Evans. Programa educativo: Simposium anemias hemolíticas. Rev Hematología. 2010 Abr-May; 11(Supl1):46-7.
2. Michel M, Chanet V, Dechartres A, Morin AS, Piette JC, Cirasino L, et al. The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood. 2009;114(15): 3167-72.
3. Kiran Dhingra K, Jain D, Mandal S, Khurana N, Singh T, Gupta N. Evans syndrome: a study of six cases with review of literature. Hematology. 2008;13(6):356-60.
4. Jin Oh H, Jae Yun M, Tae Lee S, June Lee S, Yeon Oh S, Sohn I. Evans syndrome following long-standing Hashimoto's thyroiditis and successful treatment with rituximab. Korean J Hematol. 2011;46(4):279-82.
5. Hui C, Xin-lei J, Heng-miao G, Su-yun Q. Comorbid presentation of severe novel influenza A (H1N1) and Evans syndrome: a case report. Chin Med J. 2011;124(11):1743-6.
6. Hunzelmann N. Systemic scleroderma. Ann Rheum. 2013;64(4):299-312. doi: 10.1007/s00105-013-2537-x.
7. Seung-Woo B, Myung-Won L, Hae-Won R, Kyu-Seop L, Ik-Chan S, Hyo-Jin L, et al. Clinical features and outcomes of autoimmune hemolytic anemia: a retrospective analysis of 32 cases. Korean J Hematol. 2011;46(2):111-7. DOI:10.5045/kjh.2011.46.2.111.
8. Norton A, Roberts I. Management of Evans syndrome. Br J Haematol. 2005; 132:12537. doi:10.1111/j.1365-2141.2005.05809.x
9. McLeod AG, Pai M, Carter RF, Squire J, Barr RD. Familial Evans syndrome: a report of an affected sibship. J Pediatr Hematol Oncol. 1999; 21: 2447.
10. Mathew P, Chen G, Wang W. Evans Syndrome: results of a national survey. J Pediatr Hematol Oncol. 1997; 19: 4337.
11. Savasan S, Warrier I, Ravindranath Y. The spectrum of Evans' syndrome. Arch Dis Child. 1997 Sep; 77: 2458.
12. García-Muñoz R, Rodríguez-Otero P, Pegenaute C, Merino J, Jakes-Okampo J, Llorente L, et al. Splenic marginal zone lymphoma with Evans' syndrome, autoimmunity, and peripheral gamma/delta T cells. Ann Hematol. 2009; 88(2):177-8.
13. Hauswirth AW, Skrabs C, Schutzinger C, Raderer M, Chott A, Valent P, et al. Autoimmune thrombocytopenia in non-Hodgkin's lymphomas. Haematologica. 2008;93(3):447-50. doi:10.3324/haematol.11934.
14. Ekstrom Smedby K, Vajdic CM, Falster M, Engels EA, Martínez-Maza O, Turner J, et al. Autoimmune disorders and risk of non-Hodgkin lymphoma subtypes: a pooled analysis within the InterLymph Consortium. Blood. 2008;111(8):4029-38. doi:10.1182/blood-2007-10-119974.
15. Yi Y, Zhang GS, Gong FJ, Yang JJ. Multiple myeloma complicated by Evans syndrome. Intern Med J. 2009;39:421-2.
16. Zent CS, Ding W, Reinalda MS, Schwager SM, Hoyer JD, Bowen DA, et al. Autoimmune cytopenia in chronic lymphocytic leukemia/small lymphocytic lymphoma: changes in clinical presentation and prognosis. Leuk Lymphoma. 2009;50:1261-8.
17. Michel M, Chanet V, Galicier L, Ruivard M, Levy Y, Hermine O, et al. Autoimmune thrombocytopenic purpura and common variable immunodeficiency: analysis of 21 cases and review of literature. Medicine. 2004;83(4):254-63.
18. Wang JY, Li JC, Zhu SX. One child with Evans syndrome. J Binzhou Med Univ (Chin) 2009;32:130.
19. Quinn P, Gilligan O.M, Horgan M. Evan's syndrome complicating multicentric Castleman's disease- dramatic response to rituximab. Eur J Haematol. 2004; 73:3845.
20. Kamezaki K, Fukuda T, Makino S, Harada M. Evans' syndrome following autologous peripheral blood stem cell transplantation for non-Hodgkin's lymphoma. Clin Lab Haematol. 2004; 26:291-3.
21. Urban C, Benesch M, Sovinz P, Schwinger W, Lackner H. Fatal Evans' syndrome after matched unrelated donor transplantation for hyper-IgM syndrome. Eur J Haematol. 2004; 72:444-7.
22. Schneppenheim R, Budde U, Hassenpflug W, Obser T. Severe ADAMTS-13 deficiency in childhood. Sem Hematol. 2004; 41:839.
23. Snowden JA, Martin-Rendon E, Watt SM. Clinical stem cell therapies for severe autoimmune diseases. Transfusion Medicine 2009; 19:22334. doi:10.1111/j.1365-3148.2009.00927.x
24. Kashif M, Qureshi A , Adil SN, Khurshid M. Successful use of Rituximab in Evans syndrome and refractory immune thrombocytopenic purpura. J Pak Med Assoc. 2010;60(1):64-5.
25. Garvey B. Rituximab in the treatment of autoimmune haematological disorders. Br J Haematol. 2008;141(2):149-69.
Recibido: Mayo 31, 2013
Aceptado: Julio 24, 2013
Dr. Adrián Romero González. Hospital Militar Central "Dr. Luis Díaz Soto". La Habana, Cuba. Correo electrónico: hematologia@infomed.sld.cu
