










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Oftalmología
versión On-line ISSN 1561-3070
Rev Cubana Oftalmol v.13 n.2 Ciudad de la Habana jul.-dic. 2000
Centro de Referencia Nacional de Retinosis Pigmentaria
Aspectos genéticos y clínicos del síndrome de usher
Dra. Beatriz Dyce Gordon,1 Dra. Josefina Mejías Márquez,2 Dra. Mirtha Copello Noblet,3 Dra. Raisa Hernández Baguer4 y Dra. Irma Horrach Rosa4RESUMEN
Con el objetivo de describir algunos aspectos genéticos y clínicos del Síndrome de Usher, se realizó un estudio descriptivo transversal en el Centro de Referencia Nacional de Retinosis Pigmentaria desde marzo de 1996 hasta junio de 1998, con 33 pacientes con diagnóstico de síndrome de Usher a través de la revisión de historias clínicas, entrevistas para interrogatorio y examen físico, así como para la confección e interpretación del árbol genealógico. La mayoría de los pacientes (60,60 %) presentaron el síndrome de Usher tipo II. Se encontró consanguinidad en el 29,62 % de los casos y los antecedentes patológicos familiares se observaron en 12 familias. Las manifestaciones clínicas oftalmológicas tuvieron un inicio fundamentalmente juvenil, y las audiológicas tuvieron un inicio muy precoz (congénito) en el tipo I y en la infancia, en el tipo II. En conclusión en el presente estudio, se pone de manifiesto la heterogeneidad clínica y genética del síndrome de Usher así como su carácter hereditario con patrón de herencia autosómico recesivo. Se hace necesario su diagnóstico precoz para ofrecer asesoramiento genético a los padres y poner tratamiento adecuado a las discapacidades.Descriptores DeCS: RETINITIS PIGMENTOSA; SORDERA/diagnóstico; SORDERA/prevención y control; SORDERA/terapia.
El síndrome de Usher (SU) es un heterogéneo grupo de enfermedades caracterizadas por retinosis pigmentaria (RP) y sordera sensorineural.1 La coincidencia de ambas anomalías fue reconocida por primera vez por Von Graefe en 1855 y posteriormente Charles Usher (1914-1935) corroboró su naturaleza hereditaria.2
Este síndrome constituye la primera causa de sordoceguera hereditaria y su prevalencia es variable. Oscila entre 3,0 x 100 000 en Escandinavia y 4,4 x 100 000 en Estados Unidos3 y en Cuba es la forma sindrómica de la RP más frecuente.3
Es clínica y genéticamente heterogénea y ya se conocen varios genes responsables
de éste y de las manifestaciones fenotípicas que producen.1,2 Con el objetivo de describir algunos aspectos genéticos y clínicos del SU en nuestra población, se realiza esta investigación para así profundizar en su conocimiento y contribuir a su prevención, diagnóstico y tratamiento precoz.
MÉTODOS
Se realizó un estudio descriptivo transversal sobre algunos aspectos genéticos y clínicos del SU, con 33 pacientes así diagnosticados y clasificados según la clasificación de Kimberling en 1995, 2 entre marzo de 1996 y junio de 1998 en el Centro de Referencia Nacional de RP.Los datos fueron obtenidos a través de la revisión de las historias clínicas, entrevistas y examen físico realizados a los pacientes, que luego fueron volcados en un modelo de recolección de datos. Ellos fueron: datos generales del paciente, edad de inicio de la hipoacusia, comienzo de la RP, existencia y grado de consanguinidad, tipo de Usher, antecedentes familiares de RP, de hipoacusia o de SU, así como presencia de enfermedades concomitantes.
Además, a cada familia se le confeccionó e interpretó el árbol genealógico.
Los datos fueron procesados manualmente con ayuda de una calculadora Casio DC-200 BK y se presentaron en forma de tablas y gráficos para su análisis y discusión.
RESULTADOS
La mayoría de los pacientes (60,60 %) presentaron el SU tipo II y por lo tanto hubo un mayor número de familias incluidas en este grupo. Ningún paciente presentó el tipo III de la enfermedad (fig. 1).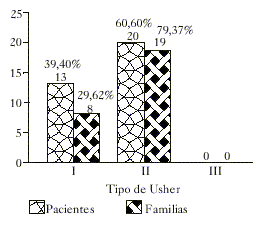
Fig. 1. Clasificación de pacientes y familias.
Se encontró consanguinidad entre los padres de los individuos afectados en el 29,62 % de los pacientes (tabla 1), y el grado de consanguinidad más común fue el de primos hermanos (75 %) (fig. 2).
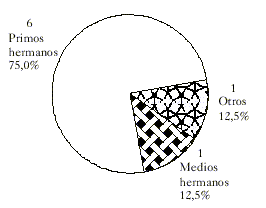
Fig. 2. Grado de consanguinidad.
| Consanguinidad familiar | | |
| Sí | | |
| No | | |
| Total | | |
| Antecedentes patológicos familiares | | |
| Retinosis pigmentaria | | |
| Hipoacusia | | |
| Síndrome de Usher | | |
| Total | | |
| Tipo | | | | ||||
| | | | | | | | |
| I | | | | | | | |
| II | | | | | | | |
| | | | |
| SU tipo I | | | |
| | | | |
| | |||
| | | ||
| | | ||
| | | ||
| SU tipo II | | | |
| | | ||
| SU tipo III | |

Fig. 3. Enfermedades concomitantes.
DISCUSIÓN
Nuestros resultados ponen de manifiesto la heterogeneidad clínica y genética del SU, al encontrar pacientes con el tipo I y II de la enfermedad, lo que está determinado por la existencia de varios genes mutantes causantes de los distintos fenotipos.2,4 Hasta el momento se han reportado los genes que se muestran en la tabla 4.Esta heterogeneidad también fue evidenciada, tanto por la diversidad de las manifestaciones clínicas como por las diferentes edades de inicio de la enfermedad. Este síndrome ha sido clasificado en 3 tipos:
SU tipo I: Caracterizado por sordera sensorineural profunda y congénita, con ausencia de la función vestibular, y RP progresiva de inicio prepuberal.2
SU tipo II: Pérdida auditiva congénita de moderada a severa y la función vestibular es normal. La RP progresiva suele comenzar alrededor de los 15 años.1,2,4
SU tipo III: Se distingue del tipo II por la naturaleza progresiva de la pérdida auditiva y el compromiso vestibular variable.2,5,6 La RP puede aparecer a cualquier edad.1
Acorde con otros reportes de la literatura7-9 el tipo II resultó ser el más frecuente, y la ausencia del tipo III posiblemente sea a causa de la rareza con que se presenta en determinadas poblaciones, estimada en el 2-4 % del total de los casos de SU.5,6
Como es de esperar para el patrón de herencia autosómico recesivo, modo de transmisión de la enfermedad,1,2 la consanguinidad tiene un papel importante en la producción de homocigotos recesivos,10 fundamentalmente entre parientes cercanos.
Conocer los antecedentes familiares en cada persona afectada contribuye a identificar el modo de herencia, pero también para sospechar la enfermedad en otros, como es el caso de los antecedentes de RP en 3 individuos con SU que deben ser valorados por el otorrinolaringólogo.
Teniendo en cuenta la naturaleza hereditaria de la enfermedad, es importante también conocer las entidades de inicio de las alteraciones oftalmológicas y audiológicas con el fin de ofrecer oportuno asesoramiento genético a los padres para que ellos decidan acerca de su futura descendencia, así como para proteger al individuo de accidentes, poner tratamiento y garantizar una educación acorde con sus discapacidades.
En la literatura 2 se reportan enfermedades asociadas al SU tales como el retraso mental y desórdenes psiquiátricos, como la esquizofrenia, la psicosis y la depresión recurrente. La psicosis fue encontrada en 1 de nuestros pacientes.
Por lo anterior se concluye que el SU es genéticamente heterogéneo. Una vez más se pone de manifiesto su carácter hereditario con patrón de herencia autosómico recesivo y la relación entre éste y la consanguinidad.
El hecho de asociar ceguera y sordera hace al individuo doblemente incapacitado, por lo que se recomienda sospechar de él siempre que se encuentre esta asociación, con el fin de realizar un diagnóstico precoz que garantice iniciar un tratamiento y manejo educacional oportunos, así como asesoramiento genético a los padres para su prevención.
SUMMARY
With the aim of describe some genetic and clinical features of Usher´s syndrome, we performed a cross and descriptive study in National Center of Remission of Pigmentosa Retinitis from March 1996 o June 1998, where 33 patients were diagnosed of Usher´s syndrome through revision of medical records, interviews for interrogation and physical examination, as well as to drawing up and interpretation of genealogical tree. Most patients (60,60 %) presenting with type II Usher´s syndrome. We found consanguinity in 29,62 % of cases and familial pathologic bacgrounds were observed in 12 families. Ophthalmologic and clinical manifestations had a youthful onset, and audiologies had a very early onset (congenital) in type I, and in infancy in type II. In conclusion, in present study was evident clinical and genetic heterogeneity of Usher´s syndrome as well as hereditary character with a autosomal recessive pattern of inheritance. It is necessary its early diagnosis to offer genetical advising to parents and to treat disabilities.Subject headings: RETINITIS PIGMENTOSA; DEAFNESS/ diagnosis; DEAFNESS/ prevention & control; DEAFNESS/ therapy.
REFERENCIAS BIBLIOGRÁFICAS
1. Mackey DA. Recent advances in hereditary disease and neuro-opthalmology. Ophthalmology 1995;6(16):48-53.
2. Kimberling WJ. Clinical and molecular genetics of Usher Syndrome. J Am Acad Audiol 1995;6:63-72.
3. Sarmiento JA. Algunas variaciones epide-miológicas de la retinosis pigmentaria en Cuba. En: Pélaez O. Retinosis pigmentaria. Experiencia cubana. La Habana: Editorial Científico-Técnica; 1997:35-47.
4. Kimberling WJ, Smith RJH. Gene mapping of the Usher syndromes. Otolaryngol Clin North Am 1992;25(5):923-34.
5. Mac Donald MI, Haney PM, Musarella MA. Summary of ocular genetic disorders and inherited systemic conditions with eye findings. Ophthalmic Genet 1998;19(1):1-17.
6. Pakarinen L, Karialainens S, Simola KO, Laippala P, Kaitalo H. Usher syndrome type 3 in Finland. Laryngoscope 1995; 105(6):613-7.
7. Herrera M. Características clínicas de la retinosis pigmentaria. En: Peláez O. Retinosis pigmentaria. Experiencia cubana. La Habana: Editorial Científico-Técnica, 1997:87- 107.
8. Hope CI, Bundey S, Proops D, Fielder AR. Usher syndrome in the city of Birmingham. Prevalence and clinical classification. Br J Ophthalmol 1997;81(1):46-53.
9. Rosenberg T, Haim M, Hauch AM, Parving A. The prevalence of Usher syndrome and other retinal dystrophy-hearing impairment associations. Clin Genet 1997;51(5):314-21.
10. Trop I, Schloss MD, Palomero R, Kaloustian V der. Usher syndrome in four siblings from a consanguineous family of Pakistani origin. J Otolaryngol 1995;24(2):102-4.Recibido: 18 de noviembre de 1999. Aprobado: 16 de diciembre de 1999.
Dra. Beatriz Dyce Gordon. Calle Cintra No. 66. entre Reyes y Empresa, Cerro, Ciudad de La Habana, Cuba.
2 Especialista de I Grado en Otorrinolaringología.
3 Especialista de II Grado en Oftalmología. Asistente.
4 Especialista de I Grado en Oftalmología.

