










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Salud Pública
versión On-line ISSN 1561-3127
Rev Cubana Salud Pública v.37 n.1 Ciudad de La Habana ene.-mar. 2011
REVISIÓN
Autoridad reguladora de medicamentos cubana. Experiencia y entrenamiento a otras autoridades latinoamericanas en la autorización de ensayos clínicos
The Cuban drug regulatory authority. Experience gained and training offered to other Latin American agencies in authorizing clinical trials
Santa Deybis Orta Hernández
Doctora en Medicina. Especialista de II Grado en Bioestadística. Investigador Auxiliar. Centro para el Control Estatal de la Calidad de los Medicamentos (CECMED). La Habana, Cuba.
RESUMEN
El Departamento de Vacunas y Biológicos de la OMS ha desarrollado una herramienta de evaluación para identificar el estado de las funciones básicas en las autoridades reguladoras y tanto la OMS como la OPS, proponen lineamientos para el entrenamiento de las autoridades reguladoras de medicamentos. La función relacionada con la autorización de ensayos clínicos ha sido desarrollada en Cuba y existe la experiencia y calificación pertinente para esta actividad. El propósito es desarrollar entrenamientos para que las autoridades de países en desarrollo, puedan implementar con éxito su función básica en la autorización de ensayos clínicos con vacunas. Se tomaron como fuentes bibliográficas, las referencias de la OMS, la Conferencia Internacional de Armonización, Unión Europea, Consejo de la Organización Internacional de Ciencias Médicas, la Administración de Drogas y Alimentos de los Estados Unidos y otros documentos reguladores de interés. Se describen los cinco indicadores que evalúan el contenido y función de las autoridades reguladoras de medicamentos, sus características y aspectos relevantes a tener en consideración en cada uno de ellos, se brinda un resumen sobre los aspectos esenciales de los estudios clínicos en vacunas según la guía de la OMS así como la experiencia de la autoridad reguladora cubana en la autorización de ensayos clínicos en vacunas y se hace referencia a las modificaciones que actualizan estos indicadores. En resumen la Autoridad Reguladora de Medicamentos es la encargada de garantizar que los medicamentos y en particular las vacunas que se administran a los seres humanos cumplan con la calidad, seguridad y eficacia, conforme los estándares y requerimientos reguladores vigentes de cada país.
Palabras clave: Centro para el Control Estatal de la Calidad de los Medicamentos (CECMED), Autoridad Reguladora de Medicamentos, autorización de ensayos clínicos con vacunas, función básica.
ABSTRACT
The Department of Vaccines and Biologics in the World Health Organization has developed an evaluation tool to identify the condition of the regulatory functions and the training of drug regulatory authorities to comply with their functions. This article was intended to provide the drug regulatory agencies from the non-developed countries with the necessary training, so that they can successfully perform their basic function in giving authorization to clinical trials with vaccines. Literature from WHO, the International Conference of Harmonization, the European Union, the Council of the International Organization of Medical Sciences, the US Food and Drug Administration and other regulatory documents of interest was reviewed. Data related to those indicators evaluating the performance of the drug regulatory authorities in giving permission for clinical trials with vaccines were collected and the five indicators assessing the contents and function of these authorities, their characteristics and relevant aspects were described. Furthermore, a summary of the essential aspects of the clinical trials with vaccines according to the WHO guideline as well as the experience of the Cuban regulatory authority in authorizing the conduction of these clinical trials was provided. The Drug Regulatory Authority is the responsible for assuring that vaccines for human beings meet the regulatory requirements of the country in the first place, and then for the research, commercialization or introduction of the vaccine in the National Immunization Program.
Key words: Center for the State Control of Drug Quality (CECMED), Drug Regulatory Authority, clinical trials with vaccines, basic function.
INTRODUCCIÓN
Para dar inicio a este artículo y lograr una clara comprensión de la complejidad y magnitud que tiene el tema, es justo mencionar algunas situaciones bien conocidas pero necesarias para fundamentar este texto. Entre otros aspectos, se encuentra el desarrollo acelerado de la industria biofarmacéutica en los últimos años, los avances en el conocimiento acerca de la prevención de las enfermedades infecciosas mediante la administración de vacunas, y la responsabilidad que tiene cada país de garantizar a la población medicamentos con una calidad, seguridad y eficacia adecuada y demostrada. Para garantizar el cumplimiento de esta responsabilidad se requiere que las Autoridades Reguladoras de Medicamentos (ARM) en los países en desarrollo cuenten con las condiciones requeridas para actuar en consecuencia.
La Organización Mundial de la Salud (OMS), a través de su Departamento de Vacunas y Productos Biológicos (V&B), asesora a la UNICEF y otros organismos de las Naciones Unidas sobre la aceptabilidad, en principio, de las vacunas que planean adquirir. El proceso seguido en la OMS para evaluar la aceptabilidad de las vacunas propuestas fue publicado inicialmente en el 39º informe del Comité de Expertos de la OMS en Patrones Biológicos (Serie de Informes Técnicos 786, anexo 1, 1989). Posteriormente fue modificado y, en 1996, sustituido por el documento Procedimiento para evaluar la aceptabilidad en principio de las vacunas a ser adquiridas por organismos de las Naciones Unidas (OMS/VSQ/97.06), en el 2003 se emite el actual documento. El sistema en vigor ha sido eficaz para promover la confianza en la calidad de las vacunas que se envían a los países a través de los organismos de adquisición de las Naciones Unidas.1
Tanto la OMS, como la OPS trabajan intensamente para lograr el fortalecimiento de las ARM´s en sus funciones básicas, que incluyen:
- Regulación Farmacéutica.
- Registro/Otras Autorizaciones de Comercialización.
- Inspecciones/Licencias a Establecimientos Farmacéuticos.
- Liberación de Lotes.
- Ensayos Clínicos.
- Análisis de Laboratorio.
- Vigilancia Poscomercialización.
El cumplimiento de todas o una parte de estas funciones, está relacionado con la producción de vacunas en los países en cuestión, de hecho los países productores, deben cumplir con todas ellas.
El Centro para el Control Estatal de la Calidad de los Medicamentos (CECMED), como ARM cubana, está certificada desde el año 2000 en el proceso de aceptabilidad para la vacuna de la hepatitis B, ha recibido inspecciones de la OMS (2000-2009), en varias ocasiones (6) y ha demostrado fortaleza en el desempeño de las funciones básicas antes referidas.
Un ejemplo de la experiencia cubana con la evaluación clínica de la autorización y modificación de ensayos clínicos, e inspecciones de Buenas Practicas Clínicas (BPC) se muestra en las tablas 1 y 2, donde se expone resumidamente, una parte del trabajo realizado en el quinquenio 2005-2009, amparado por las regulaciones vigentes.2,3
Entre otros aspectos que avalan la experiencia y el desempeño del Sistema de Evaluación de Ensayos Clínicos y Registro de Medicamentos, cuentan:
- El mejoramiento continúo de las bases técnicas y metodológicas para el desarrollo del proceso de evaluación, control e inspección.
- La magnitud, complejidad y experiencia en el proceso de la evaluación clínica para Registro y Autorización de Ensayos Clínicos.
- La implementación de un Programa Nacional de Inspecciones a Ensayos clínicos (2001).
- Formar parte de la Red de Evaluación Clínica de vacunas para países en desarrollo (DCVRN) de la OMS, desde su creación (2004).
- Certificación del INN (nombre genérico del producto) y AENOR (Institución Internacional Certificaciones ISO) del Sistema de Gestión de la Calidad (2008) y Seguimiento (2009).
- Implementación del Sistema de Certificación en BPC a sitios y servicios clínicos en el país (2008).
- Evaluación y Certificación como Autoridad Reguladora de Medicamentos de Referencia de OPS/OMS. 2010.
En septiembre del 2004, se realizó en Lima, Perú el Curso- Taller "Funciones Básicas de las Autoridades Reguladoras de Medicamentos para el Control de Vacunas" auspiciado por la OPS, con la participación de especialistas de las ARM´s de Bolivia, Colombia, Ecuador, Perú y Venezuela, como parte del entrenamiento necesario para cumplir con las funciones antes mencionadas.
En este curso se abordaron las siete funciones básicas, sin embargo en este articulo solo se refiere de manera específica a lo relacionado con la evaluación clínica de la autorización de ensayos clínicos y los indicadores establecidos para la valoración de esta función que se desarrolló en el Manual de Entrenamiento sobre funciones reguladores críticas para vacunas: Evaluación del Desempeño Clínico mediante la Autorización de Ensayos Clínicos,4 por el Departamento de Vacunas y Biológicos de la OMS en el 2003.
Se exponen a continuación las características e indicadores de esta función y su fundamento teórico-práctico, que fue impartido durante el entrenamiento.
Importancia y razón de esta función
Cabe preguntarse en este momento ¿por qué la ARM es la encargada de autorizar los ensayos clínicos? La respuesta tiene implícito dos elementos fundamentales, el primero es garantizar la seguridad, protección, derechos y beneficios a todos los sujetos que se involucran en las investigaciones biomédicas, acorde con los principios éticos internacionales, y la segunda, responde a la necesidad de garantizar que los ensayos clínicos se realicen con un diseño, conducción y análisis acorde a principios científicos, para cumplir sus objetivos.
Según plantea la OMS esta función es la encargada de garantizar que las vacunas en los seres humanos, no pueden ser investigadas, comercializadas o introducidas en el Programa de Inmunización sin primero haber reunido los requerimientos reguladores del país.
Para desarrollar este tema se tomaron referencias básicas de la OMS, el manual antes mencionado,4 así como otras referencias específicas que mencionaremos más adelante en cada uno de los indicadores y la experiencia de la ARM cubana en el establecimiento, desarrollo y cumplimiento de esta función.
SÍNTESIS DE LOS DATOS
INDICADORES
Los indicadores que evalúan la función básica, tienen dos propósitos:
- Hacer que laARM sea capaz de realizar una autoevaluación de su desempeño.
- Identificar donde son necesarias las mejoras y los entrenamientos con el fin de saber cuales son los países que requieren prioridad de entrenamiento.
Los indicadores establecidos incluyen:
1. Política de Buenas Prácticas Clínicas (BPC), Buenas Prácticas de Laboratorio (BPL), Buenas Prácticas de Manufactura (BPM) y vigilancia de los aspectos éticos en los ensayos.
2. Guías escritas que describan las condiciones bajo las cuales los ensayos clínicos serán necesitados: consideraciones emitidas para la solicitud de datos clínicos para el uso local de vacunas.
3. Pautas publicadas sobre el formato para la presentación de los datos clínicos.
4. Acceso a experticia en epidemiología y estadísticas para aconsejar sobre la ejecución y análisis de los ensayos.
5. Acceso a expertos en el producto que está siendo evaluado (incluye expertos en métodos de ensayos).1-4
A continuación se describen, en primer lugar, algunas definiciones correspondientes a cada indicador y sus generalidades para su mejor comprensión.
Indicador 1
Política de Buenas Prácticas Clínicas (BPC), Buenas Prácticas de Laboratorio (BPL), Buenas Prácticas de Manufactura (BPM) y vigilancia de los aspectos éticos en los ensayos.
Definiciones
Buenas Prácticas Clínicas. Un estándar para los ensayos clínicos que abarcan el diseño, conducción, seguimiento, auditorias, análisis, registros, información y documentación de ensayos clínicos, que asegura que los estudios son válidos ética y científicamente y que las propiedades clínicas del producto farmacéutico (diagnósticas, profilácticas o terapéuticas) bajo investigación están convenientemente documentadas.
Buenas Prácticas de Laboratorio. Un sistema de calidad relacionado con el proceso organizativo y las condiciones bajo las cuales se planifican, ejecutan, monitorean, registran, archivan e informan, los estudios no clínicos de salud y seguridad ambiental.
Buenas Prácticas de Manufactura. Es la parte del Aseguramiento de la Calidad Farmacéutica que asegura que los productos son producidos consistentemente y controlados para los estándares de calidad apropiados a su propuesta de uso y el exigido para la autorización de comercialización. (OMS)
Comité de Ética Independiente (CEI). Cuerpo independiente (un consejo de revisión, regional, nacional o supranacional), constituido por miembros médicos, científicos y no científicos, cuya responsabilidad es garantizar la protección de los derechos, seguridad y bienestar de los seres humanos implicados en un ensayo, velar por su validez científica y justificación ética y social, así como dar una garantía pública de esta protección.
Consentimiento informado. Proceso por el cual un sujeto confirma voluntariamente su disposición a participar en un ensayo determinado, después de haber sido informado y haber comprendido todos los aspectos del ensayo que son relevantes para la decisión del sujeto, incluyendo una explicación de las características de la investigación, sus objetivos, beneficios potenciales, riesgos e inconvenientes, los tratamientos alternativos disponibles y los derechos y responsabilidades del sujeto conforme la última revisión de la Declaración de Helsinki. El consentimiento estará documentado por medio de un formulario de consentimiento escrito firmado y fechado.
Generalidades
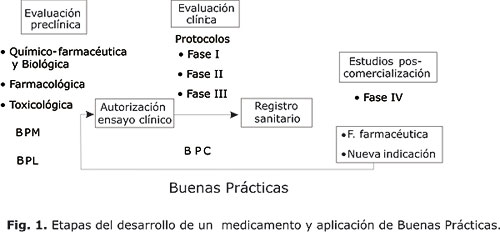
Los ensayos clínicos son operaciones complejas que demoran uno o más años e involucra a numerosos participantes en varios sitios de ensayos. Es bien conocido que para la solicitud de autorización de ensayos clínicos, las ARM´s y las organizaciones de salud pública requieren la presentación de información sobre la calidad del producto (producción, ensayos, estabilidad), estudios fármaco-toxicológicos, estándares clínicos y el protocolo, como se muestra en la figura 1.
Las ARM´s son responsables de las dos etapas de la evaluación crítica de la documentación que ampara a los estudios clínicos:
1. cuando los ensayos clínicos están siendo propuestos para solicitar autorización, y
2. cuando los resultados de los ensayos son presentados para la solicitud de autorización de comercialización.
La revisión por la ARM de la calidad del producto y de los datos preclínicos se realiza a la vez que el producto es propuesto por primera vez para uso en investigaciones con humanos. A lo largo de la realización de los estudios clínicos, si hay cambios en la fabricación o métodos de ensayos, y si se desarrollan diferentes dosis o formulaciones de la vacuna en estudio, habrán requerimientos adicionales para la ARM, con el fin de evaluar cualquier resultado de nuevos estudios preclínicos y así determinar, si ellos son suficientes para apoyar el cambio realizado.
Al respecto es esencial que la ARM tenga una básica y adecuada comprensión de los estándares de Buenas Prácticas, con el fin de determinar o buscar un consejo apropiado sobre si el producto, los estudios de soporte, los criterios de desempeño de los estudios clínicos y la protección a los sujetos, cumple con estos estándares.
Política de Buenas Prácticas Clínicas
En el campo de la aplicación de las Buenas Prácticas en general, y su extensión a los aspectos relacionados con la investigación clínica, resulta en ocasiones algo difícil comprender su concepción e incorporar sus principios básicos, pudiendo resumir al respecto, que las BPC expresan un modo de hacer las cosas, la implantación de una serie de medidas sistematizadas, con una norma internacional de calidad científica y ética, para la organización, dirección, ejecución, control, recogida de datos, documentación y verificación de los ensayos clínicos, en aras de asegurar la protección de los derechos e integridad de los sujetos involucrados en ellos, de forma tal que permitan controlar que el estudio se realice con objetivos concretos para alcanzar los propósitos con alto nivel de calidad preestablecido.
Por otra parte, es necesario reconocer lo relacionado con el empleo racional y económico de los recursos. Si un ensayo clínico se realiza con todo rigor científico y cumpliendo todas las condiciones preestablecidas, solo ha de hacerse una vez; esto es importante desde el punto de vista ético, por cuanto un menor número de sujetos serán sometidos al nuevo producto en etapa experimental. El objetivo es focalizar los esfuerzos y el éxito con estudios de mejor calidad.
Se requiere un marcado énfasis al expresar, que es precisa la aplicación y el estricto cumplimiento de las BPC en la planificación, ejecución y conducción de los ensayos clínicos, de modo que quede clara y explícita, la forma en que se aplica este concepto en toda la cadena que conforma la estrategia de evaluación clínica durante el desarrollo de un medicamento. Sus principios son aplicables a las cuatro fases de la investigación clínica de medicamentos (figura 1) incluyendo los estudios de bioequivalencia y de poscomercialización.
Han sido publicadas muchas guías y directrices de BPC por países y organismos internacionales en las que se detallan las responsabilidades de los investigadores monitores y promotores. Las Guías de BPC cubren muchos requerimientos para la realización y registro de ensayos clínicos, tales como sus prerrequisitos, justificación, protocolo escrito, aprobación de los comités de ética, consentimiento informado, aprobación de la ARM, confidencialidad de los datos de los apacientes, responsabilidades de los investigadores, promotores y monitores, contabilidad del producto en investigación (PI), manejo de datos, entre otros aspectos.
Las guías de BPC de la OMS y de la International Conference on Harmonisation (ICH), son documentos apropiados para estudiar y comprender sus principios. Ambos cubren los mismos principios pero el documento de la OMS brinda información sobre las responsabilidades y el papel de la ARM mientras que el de la ICH incluye una sesión para detallar el Manual del Investigador y un anexo con un listado de los documentos esenciales necesarios a lo largo de la ejecución de los estudios. Durante la evaluación de los ensayos clínicos, las ARM deben referirse a los estándares nacionales BPC, cuando existan y comparar con los estándares internacionales.
Política de Buenas Prácticas de Laboratorio
El sustento de los estudios no clínicos en una solicitud para autorización de ensayo clínico o comercialización, está porque son realizados de acuerdo con los estándares de BPL.
Estos estudios no clínicos son estudios en animales que investigan la seguridad y eficacia de la vacuna en modelos animales antes de su primer uso en los seres humanos. Los protocolos deben ser planificados y realizados conforme con las guías nacionales sobre la investigación animal. Los estudios de seguridad cubren las investigaciones sobre toxicología (local y sistémica), farmacocinética, metabolismo, biodistribución, toxicología reproductiva, o tumorogenicidad, según corresponda a los productos evaluados. Para la eficacia de las vacunas hay estudios de inmunogenicidad y si hay modelos animales se realizan estudios de eficacia de la protección de la enfermedad.
Los estudios de laboratorios no clínicos incluirían también el desarrollo de investigaciones in vitro o de células sobre la potencia de los lotes clínicos, para ser relacionados más tarde a la eficacia clínica, estudios de estabilidad en vida de estante de los lotes clínicos producidos más tempranamente y la validación de las condiciones de almacenamiento y embarque.
Una ARM que recibe una solicitud para autorización de comercialización para una vacuna, si no estaba involucrada en la aprobación de los ensayos clínicos de esta vacuna, deberá asegurarse, durante la revisión del dossier, que los estudios preclínicos son apropiados y adecuadamente realizados y documentados.
Política de Buenas Prácticas de Manufactura
Estas cubren el control efectivo de todos los aspectos de la fabricación, incluyendo validación y documentación de las operaciones de las instalaciones, procesos de producción, procesos de control, ensayos de control de calidad y almacenamiento, etiquetado y embalaje del producto final, pueden ser aplicadas a todos los productos medicinales, lotes de estudios clínicos hasta los lotes finales de producción comercial. Para el caso de los ensayos clínicos cualquier placebo o producto comparador debe ser producido también conforme las BPM.
Se reconoce además que las ARM de un país donde se está proponiendo realizar ensayos clínicos con un producto determinado, requerirán que la fabricación del PI cumpla con estándares de BPM apropiados a la fase del ensayo clínico. Cuando se realiza la solicitud de autorización de ensayo clínico o de autorización de comercialización, se requiere información que indique o demuestre que se han seguido las BPM.
Para el caso específico de los PI, es reconocido que la aplicación de los principios de las Buenas Prácticas es necesaria por diversas razones:
· Lograr que haya uniformidad entre los lotes del PI para, de este modo, asegurar la confiabilidad de los ensayos clínicos.
· Lograr la uniformidad entre el PI y el producto comercial y, por consiguiente, que los resultados del ensayo clínico sean pertinentes en cuanto a la eficacia e inocuidad del producto comercializado.
· Proteger a los sujetos que participan en ensayos clínicos frente a PI de mala calidad que sean el resultado de errores de fabricación (omisión de pasos críticos tales como esterilización, contaminación y contaminación cruzada, mezclas de productos, rotulado erróneo y otros) o de la calidad insuficiente de las materias primas y otros componentes.
· Documentar todos los cambios que se producen en el proceso de fabricación.
Con estos elementos, es importante conocer que aunque las principales guías de BPM son para los productos comercializados (farmacéuticos y biológicos), se han publicado por la OMS desde 1996 la Guía Buenas Prácticas de Fabricación: Directrices Suplementarias para la Fabricación de Productos Farmacéuticos en Investigación usados en Ensayos Clínicos en personas, y entre las regulaciones de la Unión Europea (UE), las guías de BPM incluyen en un anexo específico (13) sobre Manufactura de Productos Medicinales en Investigación, entre otros.
Vigilancia ética
La investigación es gobernada por acuerdos internacionales sobre principios éticos. Sin embargo la investigación en seres humanos es además revisada y aprobada por comités independientes a las investigaciones que están teniendo lugar en sus respectivas Instituciones. Estos comités son llamados Comités de Revisión Institucionales (IRB), o Comités de Ética Independientes (CEI). Esta revisión ética tiene lugar en cada sitio de investigación (en algunos países existe un IRB Central). Los requerimientos para el desarrollo de las actividades de los IRB o CEI, están descritos en las Guías de BPC y en muchas Guías de Ética independientes.
El documento base es la versión actual de la Declaración de Helsinki, una guía de los primeros principios éticos definidos por la Asamblea General de la Asociación Médica Mundial adoptada en 1964 y enmendada en 1975, 1983, 1989, 1996, 2000 y 2002, 2004, 2007. Se establece en este documento que el bienestar de los sujetos deberá prevalecer por encima de los intereses de la ciencia y la sociedad.
Los Comités de Ética no están limitados a ejercer su vigilancia solo para la aprobación inicial del protocolo de ensayo clínico y el consentimiento informado, sino que deben revisar también todos los cambios y enmiendas a estos documentos, las actualizaciones de informes anuales, auditar los datos del estudio, seguir el progreso del ensayo y obligar al investigador a reportar los informes de monitoreo, especialmente para los eventos adversos.1-14
Algunos aspectos que deben quedar claros y bien establecidos para la ARM en lo que a la vigilancia de la ética en la investigación clínica se refiere:
· la existencia de Comités de Ética formalmente definidos y establecidos en cada institución o sitio de investigación,
· la composición adecuada de los Comités de Ética, con la inclusión miembros externos referidos a la institución y
· el cumplimiento de su papel y deberes para la aprobación y supervisión de los ensayos clínicos que se ejecutan en la institución.
Indicador 2
Guías escritas que describan las condiciones bajo las cuales los ensayos clínicos serán realizados: consideraciones emitidas para la solicitud de datos clínicos para el uso local de vacunas.
Definiciones
Eficacia de vacunas (protección). Es la reducción en el cambio o riesgo del desarrollo clínico de la enfermedad después de la vacunación relacionado con los cambios o riesgo cuando no se ha realizado la vacunación. La eficacia de la vacuna mide la protección directa (ej. La protección inducida por la vacunación en la muestra de la población vacunada).
Ensayo clínico. Cualquier investigación en humanos dirigida a descubrir o verificar los efectos clínicos, farmacológicos u otros efectos farmacodinámicos de un(os) producto(s) en investigación, estudiar su absorción, distribución, metabolismo y excreción e identificar cualquier reacción adversa al producto(s) en investigación con el objeto de determinar su seguridad y eficacia.
Estudio puente. Es un estudio realizado en una nueva población para obtener datos farmacodinámicos o clínicos sobre la eficacia, la inocuidad de las dosificaciones y el régimen de administración en la nueva región que permita extrapolar el conjunto de datos clínicos foráneos.
Evento adverso. Cualquier acontecimiento médico desfavorable que se presenta en un paciente o sujeto de investigación clínica al que se administra un producto farmacéutico, y que no tiene necesariamente una relación causal con este tratamiento. Un acontecimiento o evento adverso puede ser, por tanto, cualquier signo desfavorable e inesperado (incluyendo un hallazgo de laboratorio anormal), síntoma o enfermedad temporalmente asociada con el uso de un PI, esté o no relacionado con este producto.
Immunogenicidad. Es la capacidad de una vacuna para inducir memoria inmunológica humoral (mediada por anticuerpos específicos) y celular o ambas mediante inmunidad.
Reacción adversa al medicamento. Es una respuesta a un producto farmacéutico (incluye vacunas) durante su uso clínico posterior al registro, que ocurre a dosis normalmente usadas y que fueron evaluadas en los humanos durante el desarrollo de los ensayos clínicos.
Reactogenicidad. Eventos que se considera que han ocurrido en relación causal a la medicación o vacunación. Pueden ser locales o sistémicas.
Vacunación con refuerzo o rectificación (booster: elevación de tensión). Es la vacunación dada a un cierto intervalo (al menos 6 meses) después de la vacunación primaria con el fin de inducir una protección a largo término.
Generalidades
En muchos países y regiones tienen legislados los requerimientos básicos necesarios para los ensayos clínicos, en estas regulaciones se estipula generalmente que la solicitud de autorización de comercialización de un nuevo producto medicinal (incluye vacunas) se debe acompañar de un expediente que contenga información de la producción, su control, estudios preclínicos y datos clínicos, sobre los que se especifica que deben ser obtenidos con el cumplimiento de las BPC y los estándares internacionales de la aprobación ética.
Debe quedar claro que las ARM´s deben contar con guías escritas para aquellas situaciones en que se requiera la realización de ensayos clínicos y brindarlas a los promotores y productores que desarrollan vacunas.
Los ensayos de un nuevo producto, o uno existente que sufre cambios importantes que lo hacen en efecto un nuevo producto, atraviesan las fases establecidas de los estudios clínicos que para el caso de las vacunas se muestran en la tabla 3.
Estos estudios clínicos se ejecutan de acuerdo al protocolo aprobado, y tienen lugar secuencialmente. La solicitud de los ensayos para cada fase se realiza después que se tienen los datos clínicos de la fase previa, los que se presentan para análisis y evaluación a la ARM con el protocolo clínico del nuevo estudio propuesto.
Los ensayos fase IV generalmente son estudios clínicos no controlados que se realizan en la etapa de poscomercialización, en ocasiones en condiciones similares a la fase III y recoge datos sobre la frecuencia de reacciones adversas que no pudieron ser demostrados en la fase
III o en los ensayos de efectividad de la vacuna. Los estudios de seguridad en fase IV son importantes porque involucran grandes números de personas saludables, principalmente niños y recién nacidos.
Existen sin embargo, otras situaciones donde se necesita la realización de ensayos clínicos. Pueden ser estudios parciales o repetición de pequeños ensayos para establecer la equivalencia de productos modificados o una fase específica de un ensayo para demostrar inmunogenicidad en una vía diferente de administración. Estas situaciones deben estar claramente definidas para que los productores conozcan cuando se requiere la realización de ensayos clínicos:
· Cambios significativos en el proceso de producción, para una vacuna autorizada.
· Una nueva combinación de vacunas.
· Cambios en el origen o composición del antígeno en una vacuna autorizada.
· Estudios clínicos puente para apoyar la extrapolación de los datos de seguridad, inmunogenicidad y eficacia de una población a otra.
· Cambios en los excipientes, materiales de partida o intermedios.
· Vacunas hechas por diferentes productores.
· Nuevas formulaciones, dosis. rutas de administración. indicación o población blanco.
· Nuevo adyuvante (la vacuna es la combinación vacuna/adyuvante).
· Inseguridad con respecto al cumplimiento de las BPC y revisión ética al realizar la solicitud de autorización de comercialización.
· Insuficientes datos clínicos para sustentar la propuesta.
En muchas ocasiones, depende de la individualidad de los casos, el tipo de vacuna y la situación encontrada, la respuesta y el tipo de estudio a realizar. En el caso en que una vacuna registrada la necesidad de nuevos ensayos clínicos dependerá de los cambios significativos realizados en el proceso de producción.
Por ejemplo la UE regula los cambios definiendo 2 tipos, los cambios menores, tipo I, que no afectan al producto y los cambios moderados y mayores, tipo II, que posiblemente afecten al producto. Al respecto debe tenerse cuidado pues muchos cambios que pueden ser considerados tipo I en los productos farmacéuticos, pudieran ser considerados tipo II para el caso de biológicos y vacunas, por la variabilidad inherente en la producción de biológicos. Se establece en estas regulaciones que ciertos cambios pueden alterar fundamentalmente los términos de la autorización de comercialización y en estos casos podría no considerarse una modificación sino que requeriría una nueva solicitud para proceder a una evaluación científica completa (incluyendo los ensayos) como en la etapa de obtención de Registro.
Cuando se trata de nuevas formulaciones de una vacuna, por ejemplo un nuevo excipiente o estabilizador con un perfil de seguridad conocido, la vacuna aunque sea contra la misma enfermedad, requeriría ensayos limitados para demostrar su seguridad, inmunogenicidad y eficacia y posiblemente para comparar la nueva formulación contra una original y demostrar que esta es al menos tan segura y efectiva.
Para el caso de nuevas indicaciones, si las dosis son similares, y los estudios, de fase I tienen bien probada la seguridad, entonces los estudios pudieran iniciarse a partir de la fase II, para determinar seguridad y eficacia en la población diana.
Cuando aparece una nueva ruta de administración, se necesita al menos un ensayo comparativo para demostrar que la seguridad e inmunogenicidad de la nueva ruta es equivalente o mayor que la original.
Si el cambio es en la dosis o esquema de dosis, la necesidad de realizar ensayos clínicos dependerá del cambio de dosis y de si el nuevo esquema fue estudiado en fase II o ambos. Si la dosis se reduce, se necesita al menos un estudio para evaluar seguridad e inmunogenicidad y posiblemente también se necesite un refuerzo. Si la dosis se incrementa, la necesidad de un ensayo fase I o II dependerá del conocimiento que se tenga de la seguridad de las altas dosis
Si la vacuna es propuesta para una población diana diferente (originalmente adultos y ahora en niños), entonces generalmente se requiere realizar todas las fases de los ensayos clínicos para mostrar el perfil de seguridad, los niveles de dosis, la inmunogenicidad y la eficacia.
La mayoría de los países importan vacunas que han sido estudiadas en otras partes. La mayor parte de los países industrializados aceptan datos de ensayos clínicos autorizados por otros países que cuentan con sus regulaciones y control y cumplen con los estándares internacionales. Se requiere además la valoración acerca de las características de la población en que se realizaron los estudios, para conocer si es equivalente a la población del país importador. Para estos casos se adicionan los estudios puentes diseñados para brindar información adicional que permita extrapolar los datos foráneos a la población diana local. La ARM debe decidir que datos de la vacuna original necesitan ser repetidos o si se necesita del estudio puente, para una vacuna que ha sido estudiada en otro país, en un esquema de dosis diferente.
La ARM puede decidir sobre la necesidad de aprobar o realizar estudios clínicos cuando un protocolo clínico foráneo es propuesto para estudios clínicos en el país y la propuesta no se ajusta a las necesidades del país, o a su población, o al programa de inmunización. El ensayo propuesto necesita por consiguiente ser revisado para ser relevante a la población local o el esquema de vacunación.
Con respecto a las reacciones adversas, ocasionalmente sus reportes durante los ensayos, pueden resultar en una suspensión temporal y revisión de los ensayos.
La ARM le brinda a todos los casos en que se necesita la realización de ensayos clínicos guías con requerimientos generales. El productor de la vacuna que realiza la solicitud necesitará evaluar su propio producto de acuerdo a las guías generales y entonces tomar las decisiones basado en el conocimiento de la vacuna y de la enfermedad, el plan de estudios conveniente y clasificar la solicitud en concordancia.
Se utiliza como política de muchas ARM la realización de reuniones con los productores para discutir aspectos específicos del producto.
La Evaluación Clínica de los resultados de los estudios clínicos en vacunas
Se describe en el contexto de este segundo indicador algunos aspectos, características y procederes generales de la evaluación clínica que se realiza en las ARM, conforme las normativas establecidas.
Importancia de la evaluación clínica y repercusión en el Registro Sanitario
Esta evaluación clínica tiene dos vertientes, una es la relacionada con las solicitudes de trámites de Registro (Inscripción, Renovación, Modificación, Asesoría de Investigación y Aprobación de Textos para Información de Medicamentos) y la otra es la que se realiza en el caso de trámites de Ensayo Clínico (Autorizaciones de Inicio de Ensayo Clínico y Modificación de Ensayos Clínicos Autorizados).
Caracterización de la evaluación clínica por la ARM
La evaluación clínica en todos los casos, requiere no solo de la aplicación de los conocimientos, el manejo del marco normativo nacional e internacional, la realización de revisiones bibliográficas actualizadas y el cumplimiento de los aspectos técnico-metodológicos y procedimientos establecidos para su realización, sino que además lleva implícito el análisis con profundidad y detalle de los resultados obtenidos y la experiencia, juicio y decisión de los evaluadores con el fin de emitir dictámenes que permitan en un caso garantizar la seguridad y eficacia de los medicamentos que consumirá la población y en otro, proteger adecuadamente los derechos y el bienestar de los sujetos que se involucran en los estudios clínicos.
La experiencia acumulada en el contexto de la evolución de esta actividad, incluye desde la creación y explotación de los instrumentos de trabajo (con las modificaciones correspondientes, en las diferentes etapas de desarrollo de la ARM), hasta el establecimiento de las rutinas de trabajo y el desarrollo y fortalecimiento de las bases legales, para lograr resultados satisfactorios en la misma. Han sido diversos y en ocasiones complejos, los cambios que ha tenido, para llegar a convertirse en un proceso. En la figura 2 se resume de manera esquemática lo que podría ser el proceso de evaluación por cualquiera ARM, con independencia de su estructura, desarrollo, o complejidad.
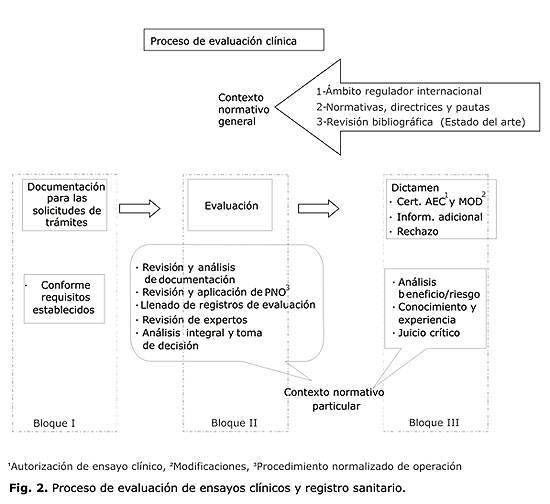
Este esquema permite abordar cada uno de los elementos relacionados con el proceso de evaluación clínica y posibilita que se muestren los resultados obtenidos en este período, con una cronología, un orden lógico y una estructura comprensible.
El primer bloque de la figura 2, abarca las características de la documentación que se presenta para trámites de ensayo clínico y registro, la que tiene que cumplir en todos los casos los requisitos establecidos por cada ARM, para poder iniciar su proceso de evaluación, teniendo en consideración que para cualquiera de los tramites a que se hace referencia, existen normativas específicas en cada país.
El segundo bloque, que se refiere a la evaluación clínica como tal, tiene en primera instancia la identificación de un contexto normativo general que resulta indispensable conocer por el evaluador para ubicarse e iniciar la ejecución de este proceso dentro del contexto normativo particular, aplicando los instrumentos y rutinas de trabajo que posibilitan su realización de manera organizada, con integralidad y eficiencia.
Este contexto normativo general, cuenta con 3 pilares fundamentales, que incluyen:
· Ámbito regulador internacional. El conocimiento y actualización sobre el ámbito regulador internacional, en el que se precisa el manejo de la actualidad internacional de las pautas, normas, guías y regulaciones de la Food and Drugs Administration (FDA, EE. UU.), de la The European Agency for the Evaluation of Medicinal Products (EMEA), ICH, Canadá, España, Inglaterra, Francia, Alemania, Nórdicos, entre otros, con énfasis particular en lo establecido por la OMS, en lo que respecta a los lineamientos y estándares en la temática relacionada con la estrategia de desarrollo y evaluación clínica de productos en investigación, en el proceso de evaluación clínica para la autorización de ensayos clínicos y la obtención del registro sanitario. Se revisa además en este contexto las normativas vigentes en los países de la región de procedencia del producto en cuestión.
· La revisión de las normativas, directrices y pautas nacionales que conforman el entorno normativo y regulador nacional, que constituyen otro de los pilares fundamentales durante el proceso de la evaluación clínica.
· Por último, no menos importante es la revisión bibliográfica que conforma el tercer pilar del contexto normativo general, y está dado por la necesidad de realizar revisiones bibliográficas específicas, en el estado del arte del tema en cuestión, lo que resulta indispensable como completamiento de la información que necesita un evaluador clínico para acometer su actividad. En esta se incluye la búsqueda para lograr un nivel de actualización sobre diferentes aspectos que pueden resultar interesantes y necesarios, teniendo en consideración en principio el tipo y características farmacológicas del PI y las indicaciones diana a las que va dirigida la terapéutica. Entre otros, se hace referencia a los antecedentes en la literatura médica de productos similares o el genérico, según proceda, el conocimiento del estado epidemiológico (incidencia, prevalencia, morbilidad, mortalidad) de la indicación blanco, la existencia de otros productos indicados como principales o alternativas en la indicación estudiada, esquemas de tratamiento, información de estudios preclínicos, antecedentes de seguridad y eficacia, si procede.
En el segundo bloque de la figura 2, bajo el término de contexto normativo particular, se agrupan los instrumentos y rutinas de trabajo que posibilitan la realización de la evaluación de manera organizada, con integralidad y evidencias documentadas de su calidad. Se ejecutan para esto, actividades tales como:
· La revisión y análisis de la documentación, que implica en primera instancia la revisión exhaustiva de la información presentada, su organización, nivel de completamiento, calidad de la presentación de los informes y el análisis detallado y profundo de los aspectos más importantes, relacionados con la acción farmacológica del producto, su acción terapéutica e indicación propuesta para estudio clínico o registro.
· La revisión y aplicación de los procedimientos normalizados de operación (PNO) para realizar las actividades inherentes a la evaluación clínica; en estos se establece la metodología para la evaluación de la información referida a la seguridad y eficacia terapéutica de los productos, que han presentado trámites en el registro y el reflejo de sus resultados en la "Información para Médicos" (hoja informativa, monografía o resumen de las características del producto) y la "Literatura Interior" (inserto o prospecto), dirigida al paciente. Para los trámites de Autorización y Modificación de Ensayos Clínicos se establece la metodología para realizar la evaluación clínica de los protocolos y para la emisión de las certificaciones y cartas con solicitud de información adicional, que avalan las decisiones tomadas y los dictámenes emitidos al concluir el proceso de evaluación.
· El llenado de los registros, es el resultado de la aplicación de los PNO, en ellos se plasma por escrito, a modo de resumen, con un formato preestablecido, la información de los diferentes aspectos relacionados con la seguridad y eficacia, evaluados en cada caso. Cada registro se corresponde con una evaluación y un trámite específico.
· La revisión de expertos, esta implícita en 2 de los indicadores de la Función Básica que trata sobre la evaluación clínica de la autorización de ensayos clínicos. Para su desarrollo se cuenta con un grupo de procedimientos que incluyen desde su reclutamiento, hasta las condiciones y características en que realizaran las evaluaciones solicitadas, ya sean independientes o formen parte de una comisión (epidemiólogos, bioestadísticos, clínicos u otros), según requieran. Los procedimientos y registros a utilizar en el proceso de evaluación se corresponden con los establecidos en el proceso de evaluación.
· Análisis integral y toma de decisión, son los elementos que casi cierran este contexto normativo particular. Para ello es necesaria la imparcialidad, credibilidad y transparencia en la toma de decisiones por lo que debe llevar implícito el análisis integral ético, científico y social que se requiere en cada caso en particular, teniendo en consideración la responsabilidad de la ARM, ante cada una de sus decisiones con respecto a la calidad, seguridad y eficacia de los productos que se comercializan en el país, así como la seguridad, protección, derechos y beneficios de los individuos que se involucran en los ensayos clínicos. Esta parte del proceso culmina de diferentes formas en cada región o país, pero en todos los casos las conclusiones deben ser colegiadas, con base científica y legal.
El tercero y último bloque de la figura 2, hace referencia al modo en que se materializan los resultados obtenidos durante el proceso de evaluación, donde se define acerca de la emisión del dictamen para cada trámite, este puede ser, la emisión de Certificados de Inscripción, Renovación o Modificación de un producto determinado o la Autorización de Inicio de un Ensayo Clínico o la Modificación de Ensayos Clínicos Autorizados, la emisión de cartas con la solicitud de Información Adicional o el Rechazo para cualquiera de los trámites ya mencionados; o la carta de respuesta a una Asesoría de Investigación.
Cualquiera que sea el dictamen que se emita, tiene explícito todos los elementos que hemos descrito y discutido anteriormente, pero además lleva implícito el análisis riesgo-beneficio que se pondera en cada uno de los productos o ensayo clínico evaluados, el conocimiento y la experiencia de los especialistas que realizan la actividad, y su juicio crítico para la posterior toma de decisiones. Esto constituye un ejemplo de cómo culmina el proceso de la evaluación clínica a los productos que solicitan los diversos trámites en el CECMED.
Sin embargo, no aparece descrito en la bibliografía, cual es y hasta donde llega la responsabilidad ética de un evaluador al emitir un juicio crítico, que permita otorgar la autorización para el inicio de un estudio clínico en el que se involucren seres humanos, valorando con estricta certeza los riesgos y beneficios que la inclusión en el estudio pueda acarrearle al individuo, la garantía de su seguridad, la oportunidad de obtener y reclamar sus derechos, y las ventajas de cualquier índole que esto puede reportarle. Esta responsabilidad es inherente a la actividad que realiza el especialista como evaluador, pero se fortalece con el conocimiento, la experiencia, el trabajo y el tiempo.
Es en este contexto que tiene lugar el análisis de la responsabilidad ética, moral y social que tiene la ARM y en particular en cada uno de los especialistas que como evaluadores desarrollan su actividad día a día. La responsabilidad que tiene la ARM para con la sociedad, está dada en varias etapas, en primer lugar durante la etapa de investigación-desarrollo de un medicamento, por la protección y seguridad al individuo que participa en los ensayos clínicos, posteriormente por la garantía de la calidad, seguridad y eficacia del medicamento que se registra para su comercialización, y en última instancia por la oportunidad del mismo, en el menor tiempo, para contribuir a darle solución a las situaciones del cuadro de morbilidad y mortalidad en cualquier sistema nacional de salud.4,5,8,15-21
Indicador 3
Pautas publicadas sobre el formato para la presentación de los datos clínicos.
Definiciones
Documentación. Todo tipo de registros en cualquier forma (incluyendo, aunque no limitado a ello, registros escritos, electrónicos magnéticos y ópticos, escáneres, rayos x, electrocardiogramas) que describan o registren los métodos y resultados de un ensayo, los factores que le afectan y las acciones realizadas.
Efectividad. Utilidad de una droga, vacuna u otro tratamiento cuando es usado por la población en un largo período, bajo condiciones no controladas en el mundo.
Efectividad de vacunas. La tasa de protección conferida por la vacunación en una cierta población. Mide la protección directa e indirecta.
Informe periódico de seguridad. Es un resumen de la información global actualizada sobre la seguridad de un producto farmacéutico, con autorización de comercialización. Se entregará a la ARM periódicamente según se establezca en el cronograma.
Protocolo. Documento que describe el objetivo(s), diseño, metodología, consideraciones éticas, estadísticas y organización de un ensayo. El protocolo habitualmente proporciona también los antecedentes y la justificación del ensayo, aunque ambos pueden quedar explícitos por otros documentos a los que haga referencia el protocolo.
Reporte de caso. Documento (que es usado) sobre los sujetos en los ensayos clínicos, en el transcurso del estudio, como está definido en el mismo. Los datos deben ser recogidos mediante procedimientos que garanticen la conservación, mantenimiento y reparo de la información y permitan el fácil acceso para la verificación, auditoría o inspección
Reporte de eventos adversos. Reporte que entrega el promotor a la ARM, de los eventos adversos procedentes de la investigación clínica en curso. El formato y tiempo para su entrega, dependerá de las normas establecidas al respecto por el país y en este debe declararse la relación de causalidad.
Generalidades
La ARM de cada país además de brindar un formato general para la presentación de una solicitud de autorización de ensayo clínico o de comercialización, debe ofrecerle a los productores instrucciones concisas sobre el formato específico para la presentación del plan de estudios clínicos (protocolo clínico) y los datos recogidos durante la ejecución del ensayo clínico (informe final). Las instrucciones para la presentación del informe anual y el reporte de reacciones adversas, se necesitan también con el objetivo de que las ARM´s permanezcan informadas sobre la seguridad clínica y garantizar que se brinda a los médicos de asistencia, la información exacta y pertinente de seguridad. Incluye:
· Formato para la sección de una solicitud para ensayos clínicos fase II, III, o IV o para autorización de comercialización.
· Reporte de eventos adversos durante los estudios clínicos o después de la comercialización.
· Actualizaciones anuales del progreso de los ensayos clínicos o actualización de seguridad.
· Formato para la información clínica sobre etiquetado (información del prospecto).
La mayoría de las ARM´s requieren que las solicitudes de autorización de ensayo clínico y de comercialización se presenten con un formato específico en correspondencia con las guías que para esto han publicado con el fin de asegurar que los méritos de cada producto sean evaluados de manera consistente y oportuna. Esto permite a la ARM, primero realizar una selección de las solicitudes que están conforme a los requisitos establecidos, segundo aceptarlas para la revisión detallada y distribución de las diferentes partes del expediente, a los evaluadores de las diferentes secciones o si es insuficiente, rechazarla.
Para las ARM´s con limitadas guías o regulaciones nacionales y para aquellas con un escaso personal y poca experiencia, la preparación de guías para la industria, es una tarea enorme.
Para ayudar en la preparación y entrenamiento de dichas ARM´s hay disponibles documentos y guías que pueden ser utilizados en ocasiones para actualizar o comparar los estándares nacionales con los internacionales. En algunos casos las ARM´s pequeñas o en desarrollo pueden elegir aceptar los formatos de una o más ARM´s bien reconocidas cuando reciban las solicitudes de los productores de estos países.
En los últimos años los miembros de la ICH han estado trabajando en la preparación de un Documento Técnico Común (CTD) para que la misma solicitud sea aceptada en Japón, la UE los EE. UU. y adoptadas por otros paìses. El CTD consta de 5 módulos (administrativo e Información para prescribir, resúmenes del CTD, calidad, informes de estudios no clínicos e informes de estudios clínicos) que cubren toda la información requerida para la autorización de comercialización. Existe además un segundo documento de la ICH (E3) que brinda información más específica sobre el contenido del informe clínico individual a ser presentado en una solicitud.
Además, las guías de ICH y de FDA para el reporte de reacciones adversas y el informe de seguridad están muy completas y actualizadas.
La información clínica que se proporciona en el prospecto y etiquetado está sujeta a la revisión y aprobación de la ARM. El objetivo es brindar información confiable y oportuna del producto a los prescriptores y usuarios. Existen un grupo numeroso de guías que estipulan la información al respecto, en particular emitidas por la FDA.
Consideraciones sobre la evaluación clínica de las vacunas
Las vacunas en su proceso de evaluación revisten características especiales que obedecen a diferentes aspectos, entre ellos:
· que son administradas a individuos saludables, mayoritariamente en población pediátrica,
· que se administran con el objetivo de prevenir las enfermedades, lo que limita la tolerancia a las reacciones adversas,
· que al contrario de los medicamentos, la acción de las vacunas es a largo plazo, tratando a menudo de proteger para la vida entera,
· que existen pocos modelos animales que permitan anticipar los efectos en los humanos,
· que son productos biológicos, altamente complejos, derivados de materia viva, que requieren ensayos y pruebas especializadas para garantizar su calidad y seguridad lote a lote. y son más susceptibles a la variabilidad,
· que la respuesta a las vacunas puede ser influenciada por factores propios del producto, el receptor y el ambiente.
Los ensayos clínicos que se realizan para la evaluación clínica de las vacunas (profilácticas) obedecen a un esquema diferente de evaluación, aunque transitan por las mismas fases (I, II, III, IV) de los ensayos que se realizan en otros tipos de medicamentos.
Los estudios iniciales se realizan para conocer la reactogenicidad, con la evaluación de la aparición de eventos adversos que se consideren relacionados con la vacunación (local o sistemática) y la tolerancia del individuo ante la vacuna.
Características de los estudios de reactogenicidad
- Evalúan la reactogenicidad de la vacuna tomando en consideración variables demográficas, clínicas y humorales (características de los grupos poblacionales, edad, sexo, raza, relación con las condiciones patológicas especiales que puedan limitar su uso, entre otros) durante la evaluación del perfil de seguridad del producto.
- Demuestran la tolerancia del producto en relación con la vía de administración, esquema de tratamiento, características de la población en estudio (riesgo, enfermedad) y otros.
- Durante el desarrollo clínico de la vacuna, en todos los estudios que se realicen deberá evaluarse la aparición de eventos adversos, de manera tal que al obtener los resultados se pueda describir detalladamente el tipo y las características observadas (clínicas y/o alteraciones de parámetros humorales) para obtener así, el perfil de seguridad de la vacuna en cuestión.
- Durante el proceso de desarrollo clínico de una vacuna, es indispensable considerar la realización de estudios en poblaciones especiales, según la o las indicaciones del producto, el tipo de población (niños, gestantes, enfermos, ancianos) y el esquema posológico a administrar, en la evaluación de reactogenicidad.
- Para el caso de vacunas combinadas se realizarán estudios que demuestren similar o menor perfil de seguridad de la vacuna en estudio, en comparación con el de las vacunas por separadas, pero administradas simultáneamente.
Otro tipo de estudio que se realiza durante la evaluación clínica de las vacunas son los de inmunogenicidad, en los que se mide la respuesta inmune del individuo, la calidad y permanencia de esta respuesta, en términos de eficacia terapéutica. Para el caso de las vacunas es indispensable la evaluación farmacodinámica de la respuesta inmune, así como la seroconversión, seroprotección y la duración de la respuesta.
Características de los estudios farmacológicos con vacunas
Se evalúan los efectos farmacodinámicos al conocer sobre:
a) Las características de la respuesta inmune de acuerdo al conocimiento o supuesta actividad de la vacuna evaluada
· niveles,
· clases, subclases y
· función del anticuerpo especifico producido.
b) El tiempo de aparición y duración de los títulos obtenidos.
c) La inducción de la inmunidad producida por células.
d) La formación de anticuerpos neutralizantes.
e) La reacción cruzada de anticuerpos.
f) La formación de inmunocomplejos.
g) Interacciones que puedan afectar al sistema inmune
Características de los estudios de inmunogenicidad
La inmunogenicidad se reconoce como la capacidad de una vacuna para inducir inmunidad, ya sea, para respuesta humoral (por anticuerpos específicos y/o mediados por células). En estos estudios:
a) Se evalúa la respuesta serológica (seronegativos y seropositivos) de la vacuna en estudio.
b) Se determinan las dosis óptimas a administrar. Deben realizarse ensayos dosis/respuesta, haciendo énfasis en la evaluación del intervalo óptimo entre las primeras inmunizaciones y las dosis de refuerzo, cuando proceda.
c) Se evalúa la inmunogenicidad relacionada con la vía de administración, dosis, características de la población en estudio (riesgo, enfermedad) y en el caso de poblaciones especiales (niños, gestantes, ancianos) cuando proceda.
d) Se mide la seroconversion, midiendo los niveles de anticuerpos y/o seroprotección, con diseños de estudios que evalúan los valores numéricos de los títulos y la cualidad de los mismos, teniendo en consideración la afinidad, funcionalidad, epítope
de reconocimiento y otros parámetros importantes en la determinación de la calidad de una respuesta de anticuerpos.
e) Se realizan estudios de reto acorde con el tipo de vacuna (de acuerdo con las características de los patógenos en evaluación, ej. cólera).
f) Se debe demostrar para el caso de vacunas combinadas, que la inmunogenicidad inducida por la combinación, comparada con la inducida por separado en cada uno de los componentes, pero administradas simultáneamente, es similar. Se evalúa la inmunogenicidad de cada serotipo o componente presente en la combinación. Se debe evaluar también la interferencia inmunológica.
Aspectos particulares de los ensayos clínicos con vacunas
a) Los estudios controlados deben demostrar que la respuesta inmune es atribuible a la vacuna en estudio y los estudios no controlados, apoyan y fortalecen los resultados de la inmunogenicidad.
b) Siempre los ensayos clínicos en vacunas combinadas deben evaluar y demostrar las ventajas terapéuticas de la asociación en relación con el uso individual de las vacunas por separado.
c) Los estudios de poscomercialización se realizan para monitorear el desempeño de la vacuna en la población indicada bajo las condiciones de rutina de su uso. Su objetivo es detectar reacciones adversas raras o inesperadas y monitorear la eficacia y la efectividad de la vacuna.
d) La efectividad de vacunas mide la protección directa e indirecta (ej. protección de los individuos no vacunados por la inmunidad de la población vacunada). Está además determinada por la cobertura de la vacunación, la correlación de las cepas de la vacuna con las cepas circulantes y la selección de cepas no incluidas en la vacuna seguido a la introducción de la vacuna en esta población.17,18,19-25
Indicador 4
Acceso a experticia en epidemiología y estadísticas para aconsejar sobre la ejecución y análisis de los ensayos
Definiciones
Brote. Es la ocurrencia de dos o más casos ligados a una enfermedad transmisible.
Cluster. Es la ocurrencia de un conglomerado inusual de casos que se produce en personas, lugar o tiempo y se utiliza como grupo en un estudio.
Estudio caso-control. Es un estudio observacional en el que las personas con una determinada enfermedad (casos) son comparadas con otras que no presentan la enfermedad (controles-vacunados), en cuanto a exposiciones previas a factores de riesgo.
Estudio de tasa de ataque secundaria. Una investigación de un brote en una población susceptible definida. La población a ser estudiada es o un cluster (en un asentamiento urbano o suburbano) o una local (o familia). Las investigaciones de los brotes pueden ser observacionales o experimentales.
Estudios observacionales. Estudio epidemiológico que se focaliza sobre eventos, exposiciones y enfermedades que ocurren en la población durante el curso de la rutina de la vida diaria. No hay intervención experimental sobre el individuo.
Prevalencia. El número de personas que tiene una enfermedad a un tiempo específico.
Seroconversion. Incremento predefinido en la concentración de anticuerpos, considerado para correlacionar con la transición de seropositivo a seronegativo, brinda información de la inmunogenicidad de una vacuna. Si hay anticuerpos pre existentes la seroconversión es definida por una transición de una desprotección clínica a un estado de protección.
Vacunas combinadas. Producto diseñado para proteger contra:
1.una compleja enfermedad infecciosa causada por diferentes cepas o serotipos de un organismo,
2. o protección contra múltiples enfermedades infecciosas o
3. la combinación de 1 y 2.
Variable serológica subrogada. Concentración predefinida de anticuerpos correlacionada con la protección clínica.
Generalidades
Los productores gastan una gran cantidad de tiempo y dinero en el proceso de desarrollo de una vacuna para obtener la aprobación de un producto que sea comercialmente viable.
Se reconoce que aún las ARM´s bien establecidas pueden no tener necesariamente suficiente experticia en epidemiología y bioestadística, como para llevar a cabo una buena y confiable evaluación de cada una de las vacunas que se solicitan, especialmente en el caso de las nuevas vacunas y para el caso de las ARM´s pequeñas o en desarrollo puede que no tengan ningún personal con experticia específica en epidemiología y bioestadística capaz de evaluar críticamente cada vacuna que se presenta a ensayo clínico o comercialización.
La mayoría de las ARM´s pueden necesitar contratar expertos en la enfermedad que está siendo estudiada, expertos en el diseño de ensayos clínicos en vacunas y bioestadísticas para:
a) aconsejar sobre los propósitos del ensayo clínico que está siendo presentado para aprobación en el país,
b) revisar y evaluar los ensayos realizados en el país en cada fase para autorizar la próxima fase y
c) evaluar los datos y pretensiones del informe final de los ensayos clínicos concluidos en el país o en el extranjero, en las solicitudes de autorización de comercialización
Las ARM´s pueden necesitar diferentes expertos para diferentes vacunas, especialmente en epidemiología. Sin embargo un bioestadístico puede ser capaz de evaluar si el plan y los métodos estadísticos son convenientes para el diseño del estudio de la vacuna propuesta.
Cada ARM debe tener al menos un nivel básico de comprensión para estos tópicos, así como ser capaces de elegir apropiadamente sus expertos y entender y discutir los detalles de los informes preparados por ellos.
Debe quedar claro que cuando los expertos son contratados como evaluadores, ellos no eximen a la ARM de la responsabilidad para tomar la decisión final sobre la aceptabilidad de los datos y la información contenida en la solicitud. Se identifica aquí la necesidad de realizar un esfuerzo colaborativo entre los expertos y la ARM.
Una vez recibida y examinado el formato, cantidad, calidad y completamiento de la documentación de un protocolo o informe final de un ensayo clínico, la ARM tendría que reclutar el experto apropiado (preferiblemente de un Comité de Expertos Clínicos), planificar como será transmitida la confidencialidad de los datos al experto evaluador, acordar sobre el tiempo de revisión y planificar la reunión para discutir los hallazgos con el experto.
Es necesario e indispensable que se le brinde al experto una guía sobre la evaluación que debe realizar. La ARM le proporcionará un esqueleto de la información que necesita de los expertos, las check list (listas de chequeo ) de los aspectos importantes a ser evaluados y las conclusiones a emitir y le brindará además, un resumen de la calidad y los estudios preclínicos de la vacuna, así como cualquier literatura o bibliografía sobre la enfermedad o la vacuna que se haya presentado con la solicitud. Cuando concurra más de un experto, en la evaluación de una vacuna, la ARM se reunirá con todos para discutir los hallazgos. El personal de la ARM debe prepararse para la realización de estas actividades.
Hay muchos tipos de ensayos que pueden ser utilizados para estudiar las vacunas en sus diferentes estadios de desarrollo. Se resume a continuación en la tabla 4, una relación de situaciones a evaluar en correspondencia con los tipos de estudios a realizar en las cuatro fases de los ensayos clínicos.
La OMS en su Guía para la Evaluación Clínica de Vacunas: Expectativa Reguladora,5 brinda un resumen sobre las características de los estudios clínicos en vacunas, atendiendo a las fases de desarrollo, e incluye los objetivos principales o propósitos de los estudios, los métodos de evaluación o diseños máas utilizados y la población a la que van dirigidos los ensayos, información que se expone en la tabla 5, con un formato que facilita la comprensión de la información y de los resultados obtenidos en el proceso de evaluación clínica de una vacuna.
Tabla 5. Características de los ensayos clínicos en vacunas, según fase, propósitos, métodos y población a incluida
| Fase | Propósitos | Métodos | Población |
| I | Evaluación de seguridad. | Tolerancia: | Adultos sanos. |
| II | Expandir evaluación de seguridad. | Estudios abiertos, comparativos. | Adultos y niños sanos de bajo riesgo. |
| III | Eficacia protectora. | Estudios aleatorizados bien controlados (ej. placebo control). | Usualmente población de alto riesgo para enfermedades de baja incidencia |
| IV | Estudios experimentales. Estudios observacionales. | Ensayos prospectivos aleatorizados. | |
Es importante, el establecimiento y firma de los acuerdos de confidencialidad con los expertos evaluadores externos. Las ARM´s deben tener certeza de que los potenciales expertos no tienen conexión con los productores o competidores productores de vacunas, para evitar un real o aparente conflicto de intereses.
El diseño elegido por el productor y el número de sujetos incluidos estará en dependencia del tipo de vacuna, la enfermedad, la prevalencia en la población y la inefectividad de la enfermedad.
Cuando existe información de la vacuna a evaluar, se pueden requerir consejos limitados de los expertos, y generalmente estas solicitudes obedecen a modificaciones en los productos, cuyas características mencionamos con anterioridad (Indicador 2). Los requerimientos de los ensayos clínicos dependerán de la situación que se presente, el tipo de estudio a realizar, la etapa de desarrollo de la vacuna en estudio y otras consideraciones que pueda tener la ARM al respecto.
Siempre que se trate de las fases iniciales de estudios de una nueva vacuna en el país, o una solicitud de autorización de comercialización de una nueva vacuna para la cual los ensayos fueron realizados fuera del país, la ARM necesitara una profunda revisión de los expertos.17,20,23,24,26-28
Indicador 5
Acceso a expertos en el producto que está siendo evaluado (incluye expertos en métodos de ensayos)
Definiciones
Producto comparador. Un producto farmacéutico u otro producto (que puede ser placebo) utilizado como referencia en un ensayo clínico.
Control. Cualquier comparador adecuado para la validación del ensayo clínico. El comparador puede ser un tratamiento activo o un control placebo.
Placebo. Un comparador en un ensayo de vacunas, que no incluye el antígeno bajo estudio. En el caso de estudios con vacunas monovalentes, debe implicar placebo inerte (solución salina, vehículo de vacunas, etc.) o una vacuna antigénicamente diferente. En las vacunas combinadas, esto puede implicar que un brazo control en el estudio, no se le aplique tratamiento con vacuna.
Potencia. La medida cuantitativa de la habilidad o capacidad específica del producto para alcanzar un efecto biológico definido.
Generalidades
En todo ensayo clínico hay dos tipos de productos involucrados, uno es la vacuna a administrar y el otro son las muestras (sangre, suero, orina), que son recogidas durante el ensayo clínico y evaluadas para determinar los parámetros de seguridad y variables de eficacia.
Aunque generalmente para las vacunas los parámetros de seguridad usuales son las reacciones locales y sistémicas, en algunos estudios clínicos se incluyen muestras de sangre dependiendo de las reacciones esperadas al producto. La evaluación de inmunogenicidad incluye la toma de muestras de sangre o suero y la evaluación de eficacia incluye pruebas en heces para evaluar los virus.
En este indicador se tratan los aspectos específicos a tener en consideración para la evaluación del PI y las muestras relacionadas con su evaluación, por lo que se requiere que si la ARM no tiene la experticia suficiente se utilicen expertos en la materia para realizar dichas evaluaciones. Se aborda tanto para la evaluación en el proceso de autorización de ensayo clínico, o para cuando se trata de informes finales una vez concluidos los estudios, ya sea con el fin de continuar los ensayos clínicos en otras fases o para la comercialización.
Cuando el producto es la vacuna en estudio
Para el PI está bastante clara la importancia de que este sea producido conforme los requisitos estándares para la fase del ensayo que se ejecutará, como se discutió en el indicador 1.
Si se utiliza placebo en el ensayo este debe responder a los estándares de calidad del producto en estudio. Si se utiliza un producto comparador en el ensayo (la misma vacuna de otro productor o la misma vacuna con características diferentes), será necesario conocer si está registrado en el país donde se ejecuta el estudio y puede ser administrado de acuerdo a las instrucciones aprobadas o si hay necesidad de aprobar el producto comparador. Esto puede ser en ocasiones más complicado si se trata de un estudio multipaíses y el producto comparador no es idéntico. Puede suceder que la propia vacuna comparadora sea una vacuna combinada registrada.
Otra característica importante a evaluar es la estabilidad de la vacuna y del placebo (si es relevante). Además deben ser considerados el embarque, la distribución y almacenamiento en el sitio del ensayo clínico, así, como el almacenamiento y manejo por el investigador clínico y los que administran las vacunas. Es importante conocer que todos los lotes que se utilizan en el ensayo cumplen todas las especificaciones y que todos los productos utilizados (vacuna, placebo, comparador) son bien manejados.
Deben estar disponibles para la revisión por el personal de ARM u otros, los resultados de las pruebas de potencia e inmunogenicidad de cada lote de vacuna utilizada en el ensayo y el seguimiento de las temperaturas de refrigeración durante el embarque y la distribución.
Si no existe, o no está disponible la experticia en la ARM, para evaluar estos aspectos señalados, deberá contactarse con expertos que puedan aconsejar al respecto.
Cuando el producto son las muestras de los pacientes (de cada participante en el ensayo)
Para emitir el juicio final sobre la seguridad, inmunogenicidad y en ocasiones sobre la eficacia es necesario contar con los resultados de las pruebas realizadas a los pacientes durante los ensayos clínicos, por esta razón es importante y en especial en los estudios multicéntricos la revisión cuidadosa para asegurar que las muestras han sido recogidas de acuerdo al esquema aprobado que se han tenido en cuenta cualquiera de los requisitos para su preparación o extracción y que han sido testada conforme los métodos aprobados, por un laboratorio calificado, usando métodos validados y equipos calibrados.
Estas normas deben cumplirse en cualquier laboratorio en que sean procesadas las muestras, y existir en ellos los hechos que demuestren la adhesión al protocolo y cumplimiento de las Buenas Prácticas.
Los aspectos importantes a tener en cuenta al respecto se abordan conforme se trate de la evaluación de una solicitud de ensayos clínicos o de los informes finales de los estudios clínicos, para la solicitud de autorización de comercialización.
Revisión para la solicitud de ensayo clínico, por la ARM
a) Indicación clara en el protocolo de las muestras que deben ser tomadas, el esquema de muestra, los intervalos de tiempo aceptables para cada muestra, el método para la toma de muestras, el envase a utilizar, el etiquetado, la preparación de las muestras, las condiciones de almacenamiento y el tiempo límite para su procesamiento. Incluye además los procedimientos para la seguridad de la recogida de las muestras, transporte y procesamiento.
b) Indicación de los valores normales de laboratorio.
c) Detalles de los parámetros clínicos que van a ser medidos en los pacientes: temperatura (oral, rectal, axilar, duración, tipo de termómetro), signos vitales, peso, talla, química sanguínea, entre otras.
d) Especificación del método para procesar las muestras de los pacientes.
e) Especificar que laboratorios van a realizar las pruebas (central o externo) y asegurar que los resultados son equivalentes. Especificar que pruebas serán realizadas en cada laboratorio.
f) Conocer si el (los) laboratorio(s) elegido(s) para la realización de las pruebas, está(n) acreditado(s).
g) Cuando existan diferentes métodos en diferentes sitios se debe determinar de antemano como van a ser comparados los resultados.
h) Asegurarse de que en el Reporte de Caso del médico o la enfermera, quedan claras las instrucciones para la toma de muestras, almacenamiento y otras.
i) Revisar las pruebas propuestas para las muestras de los pacientes y asegurar que son apropiadas para los parámetros que van a ser medidos.
j) Acceso a expertos en la enfermedad y en los tipos de muestras tomadas para asegurar que los métodos de prueba son apropiados a las muestras tomadas.
k) Para el caso de los ensayos comparativos y en dependencia del diseño del ensayo, las pruebas realizadas a las muestras de los pacientes deberán tener suficiente sensibilidad para distinguir superioridad y no inferioridad de las vacunas preparadas.
l) Revisar, para los análisis de laboratorio, si existe control de la calidad o un sistema de aseguramiento de la calidad que haya validado los resultados.
Revisión de los informes finales de los ensayos clínicos
Tanto si se tratan de los estudios concluidos de fase I y II en una solicitud para ensayos clínicos de una fase superior o en una solicitud de autorización de comercialización. Evaluar si:
a) El informe final muestra que hubo adherencia al protocolo y se declaran y discuten las desviaciones en la toma de muestras y su procesamiento.
b) Los registros demuestran la adhesión al protocolo con respecto al etiquetado de las muestras y su preparación, las condiciones de almacenamiento y el tiempo límite para su procesamiento.
c) Se indica en el informe los valores normales de laboratorio y se destacan los valores anormales en cada paciente.
d) Se especifican en cada paciente los laboratorios que realizan las pruebas.
e) Son comparables los resultados que se obtienen, cuando las pruebas se realizan en diferentes laboratorios, por diferentes métodos.
f) Los registros de los Reportes de Caso incluyen los datos sobre la toma de muestras, almacenamiento, entre otros.
g) Se revisaron las pruebas propuestas para las muestras de los pacientes y se aseguró que eran los parámetros apropiados para ser medidos.
h) Se utilizó la evaluación de expertos en la enfermedad y en los tipos de muestras tomadas para asegurar que los métodos de prueba son apropiados.
i) Se evaluó si las pruebas realizadas a las muestras de los pacientes tenían suficiente sensibilidad para distinguir superioridad y no inferioridad entre las vacunas comparadas, en los ensayos en que procede (comparativos, u otros).
j) Si existen evidencias de cumplimiento de BPC.
Existe un control de la calidad o un sistema de aseguramiento de la Calidad que haya validado los resultados de los análisis de laboratorio realizados en el ensayo.5,17,19,29-33
Aún cuando hemos dado una explicación detallada de esta herramienta creada hace más de un quinquenio, la experiencia en las evaluaciones realizadas por OMS y OPS, el desarrollo de las ARM´s en sus diferentes funciones básicas y los resultados de los entrenamientos, han permitido mejorar estas herramientas y actualizarlas en correspondencia con sul desempeño.
Finalmente se puede concluir que la Autoridad Reguladora de Medicamentos es la encargada de garantizar que los medicamentos y en particular las vacunas que se administran a los seres humanos cumplan con la calidad, seguridad y eficacia, conforme los estándares y requerimientos reguladores vigentes de cada país, tiene cada día nuevos retos, que responden entre otros aspectos a la agilización de los procesos de evaluación, lo que implica un refuerzo del sistema de Registro y de farmacovigilancia. Este tema será abordado en próximos artículos.
REFERENCIAS BIBLIOGRÁFICAS
1. Procedimiento para evaluar la aceptabilidad, en principio, de las vacunas que vayan a ser adquiridas por organismos de las Naciones Unidas. Ginebra OMS, Vacunas y Productos Biológicos. Washington, D.c.: WHO/V&B/02.08.; 2003.
2. Regulación No. 21-2008. Requisitos para la Autorización y Modificación de ensayos clínicos. La Habana: CECMED; 2008.
3. Regulación No. 52-2008. Requerimientos para la Certificación de Buenas Prácticas Clínicas. La Habana: CECMED; 2008.
4. Training manual on the critical regulatory function for vaccines: evaluation of clinical performance trough authorized clinical trials. Geneva: WHO; 2003.
5. Guidelines on Clinical Evaluation of Vaccines: Regulatory Expectations. Geneva: WHO; 2002.
6. Good Clinical Practices. Document of the Americas. Pan American Network for Group Regulatory Harmonization. Washington, D.C.: PAHO; 2005.
7. Consejo de la Organización Internacional de Ciencias Médicas (CIOMS). International ethical guidelines for biomedical research involving human subjects. Geneva: CIOMS; 2002.
8. Handbook for good clinical research practice (GCP): guidance for implementation. Geneva: WHO. 2005.
9. Good manufacturing practices for biological products. Annex 1. [WHO Technical Report Series No. 822]. Geneva: WHO; 1992.
10. Good manufacturing practices. Vol 4. Manufacture of investigational medicinal products. Annex 13. Brusellas: European Commission; 2003.
11. E6 Good Clinical Practice: Consolidated Guidance. London: ICH;1996.
12. Regulación No. 37-2004. Buenas prácticas de laboratorio para el control de medicamentos. La Habana: CECMED; 2004.
13. Ethical principles for medical research involving human subjects. Declaration of Helsinki. Tokio: World Medical Association; 2004.
14. Resolución MINSAP 165 Directrices sobre Buenas Prácticas Clínicas. La Habana: CECMED; 2000.
15. Q9 Quality Risk Management. Guidance for Industry. Rockville: International Conference on Harmonisation (ICH); 2006.
16. Q8 Pharmaceutical Development. Guidance for Industry. Rockville: International Conference on Harmonisation (ICH); 2006.
17. E3 Guideline for Industry. Structure and Content of Clinical. Study Reports. Rockville: ICH; 1996.
18. FDA. Concerning demonstration of comparability of human biological products, including therapeutic biotechnology-derived products. EE.UU.: FDA; 1996.
19. Review of existing documents on planning, performance and assessment of clinical studies on vaccines. Geneva: WHO; 1999.
20. Surveillance of adverse events following immunization. Field Guide for managers of immunization programmes. Geneva: WHO; 1997.
21. Clinical Trial Unit Standard Operation Procedures for Clinical Trials. London (UK): Medical Research Council; 2002.
22. Real Decreto 223/2004, por el que se regulan los ensayos clínicos con medicamentos. (BOE No. 33). Madrid: Editorial Aranzadi S.A.; 2004.
23. Guidance for Clinical Trial Sponsors: Establishment and Operation of Clinical Trial Data Monitoring Committees. Washington, D.C.: FDA; 2006.
24. FDA. Content and format of chemistry, manufacturing and controls information and establishment description information for a vaccine or related product-guidance for industry. EE. UU.: FDA; 1999.
25. FDA. Postmarketing safety reporting for human drug and biological products including vaccines. EE. UU.: FDA; 2001.
26. Guidelines for the evaluation of dengue vaccines in populations exposed to natural infection. Geneva: WHO; 2001.
27. Protocols for the evaluation of epidemiological surveillance systems. Geneva: World; 1997.
28. Points to consider on the development of live attenuated influenza vaccines. London: Unión Europea; 2001.
29. Guidelines for the production and quality control of synthetic peptide vaccines. [WHO Technical Report Series No. 889]. Geneva. WHO; 1999.
30. Good practice in relation to supplies for publicly funded clinical trials of medicines. London (UK): Medical Research Council; 2004.
31. Regulation of vaccines: building on existing drug regulatory authorities. Geneva: WHO; 1999.
32. Recommendations for the production and control of Haemophilus influenzae type b conjugate vaccines. [WHO Technical Report Series No. 897]. Geneva: WHO; 2000.
33. Recommendations and guidelines for biological substances used in medicine and other documents. Annex 3. [WHO Technical Report Series No. 897]. Geneva: WHO; 2000.
Recibido: 28 de enero de 2010.
Aprobado: 5 de marzo de 2010.
Santa Deybis Orta Hernández. Responsable del Proceso de Autorización de Ensayos Clínicos en el CECMED.Centro para el Control Estatal de los Medicamentos (CECMED). Calle 200 No. 1706 e/ 17 y 19. Siboney, Playa 11600. La Habana, Cuba. Correo electrónico: deybes@infomed.sld.cu


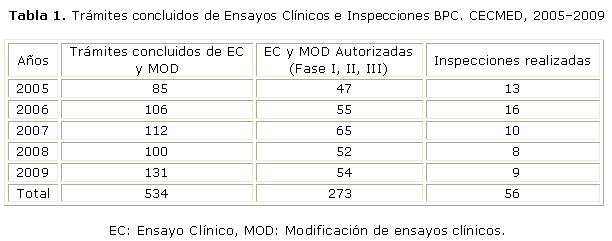{kind=link}
{kind=link}
{kind=link}
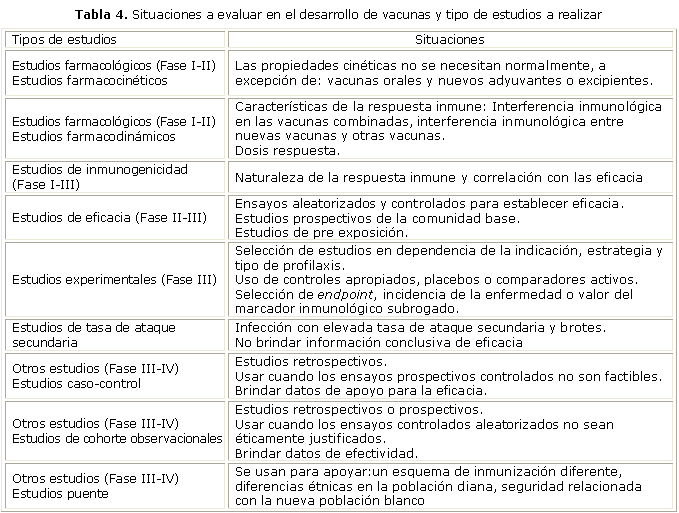{kind=link}